


You are here: Home > Section on Clinical and Developmental Genomics
Genetic and Genomic Studies in Normal Development and Diseases

- Owen M. Rennert, MD, Head, Section on Clinical Genomics
- Vanessa Baxendale, BS, Laboratory Manager
- Catherine Boucheron, PhD, Postdoctoral Fellow
- Wai Yee Chan, PhD, Adjunct Scientist
- Albert Cheung, BS, MS, Graduate Student
- Lawrence Cho, Summer Student
- Jessica Clark, BS, Postbaccalaureate Fellow
- Elizabeth Jiali Fang, Summer Student
- Tin Lap Lee, PhD, Staff Scientist
- Dalton Liu, MS, PhD, NIH/Chinese University of Hong Kong Graduate Student
- Sohan Nagrani, BS, Postbaccalaureate Fellow
- Alan Lap-Yin Pang, PhD, Biologist
- Margarita Raygada, MS, PhD, Staff Genetic Counselor
- Mark Ziats, BS, NIH/Oxford/Cambridge and Medical Scientist Training Program Graduate Student
The Laboratory of Clinical and Developmental Genomics uses genetic and genomic technologies, acquired through basic studies, to solve clinical and fundamental biological problems. An additional mission is to train physicians and postdoctoral scientists in the application of the genomic approaches to the study of human disease. One of our recent investigations attempted to define the regulatory pathways of human spermatogenesis and oogenesis, employing methodologies such as methylation tiling array to study transcriptional regulation of mArd2, a novel testis-specific gene that was first cloned in this laboratory. We also studied the role of Lin 28, a gene that regulates developmental timing, employing proteomic approaches to identify RNAs bound to the encoded protein. Gene knockdown experiments are under way to study the function of Lin 28 in determining cell fate. We also continued our studies to identify and screen for risk factors of complex disorders. In addition to fundamental and translational research, we are involved in clinical protocols to study patients with genetic and malformational disorders, and we recently initiated novel investigational approaches to study childhood autism. This spectrum of investigational approaches affords us the opportunity to provide clinical genetics training to our trainees and clinical fellows.
Effect of vitamin A deficiency on epigenomics of male germ cells
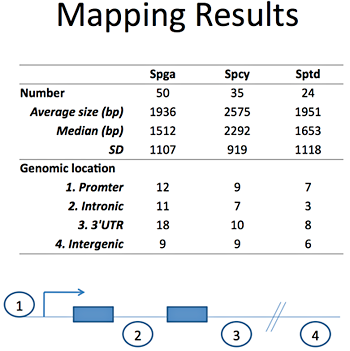
Figure 1. List of predicted lncRNA candidates
Spga, spermatogonia; Spcy, spermatocytes; Sptd; spermatids
First, we continued our description of the mouse model of vitamin A deficiency (VAD) by studying the effects of the VAD diet, from 7 weeks up to 28 weeks. For each time point, we assessed the vitamin A status of the animal: the RBP (retinol binding protein) level in the serum, and the mRNA expression levels of retinoic acid (active metabolite of vitamin A) receptors in the liver, brain, and testis. In parallel, we used histological techniques to characterize the effect of VAD on the testis of these animals. Our data showed that the number of differentiated germ cells began to decline around 16 weeks of diet, coinciding with an the appearance of an increasing number of apoptotic cells.
In the second part of our work, we employed expression profiling to study the genetic mechanism of vitamin A deficiency–induced arrest of spermatogonial stem cell differentiation. We isolated enriched populations of spermatogonia by the STAPUT method (sedimentation velocity at unit gravity in a 2%–4% BSA gradient), using microarray analysis to compare the gene expression levels of these populations. We compared gene profiles in cells from controls with those of mice fed VAD for 18 and 25 weeks; we identified 1900 differentially expressed genes in the 18-week VAD group, and 9987 in the 25-week VAD group. To identify clusters of genes and/or signaling pathways known to play a role in the regulation of spermatogenesis and which may be affected by VAD, we then carried out a more comprehensive bioinformatics analysis of control spermatogonia and those obtained after 25 weeks of VAD. Our initial analyses focused on the retinoid signaling pathways, which verified its hypoexpression in spermatogonia of the vitamin A–deficient mice. Our data also highlighted the importance of other pathways such as NF-κB, thyroid hormones, cell cycle, and lipid metabolism. Lastly, we found effects of vitamin A on cytoskeleton remodeling and cell adhesion (including cell junctions between germ cells and somatic cells).
In summary, we observed the following effects of VAD on adult mouse spermatogenesis: arrest of germ cell differentiation, increased germ cells apoptosis, and a general effect on the transcriptome profile of spermatogonia. While these effects on germ cells were sometimes dramatic, our data point to the importance of the VAD-induced effects on the somatic cells, and their indirect effects on the progression of spermatogenesis.
Effect of vitamin A deficiency on Sertoli and Leydig cells
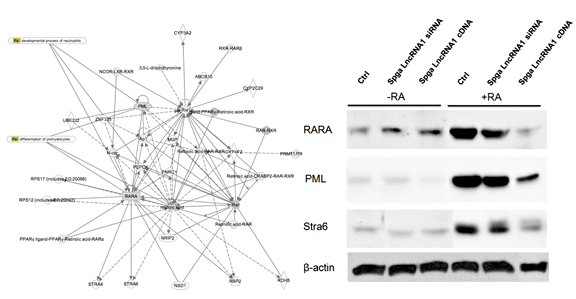
Figure 2. Effect of Spga lncRNA candidates on differentiation targets in the retinoic acid regulatory gene network, using the p19 cell differentiation model
Ctrl, control; Spga, spermatogonia. (click image to enlarge)
The results described above on the effects of VAD on germ cells suggest that this diet may also induce a deregulation of the number and differentiated stage of somatic cells (Sertoli and Leydig cells). Our hypothesis is that VAD would induce a proliferation of these cells and a consequent regression into an undifferentiated stage. Sertoli and Leydig cells would therefore become unable to regulate and sustain the proliferation and differentiation of the germ cells, which would contribute to the VAD-induced arrest of spermatogenesis. We therefore expanded our study to the observation of these somatic cells by verifying potential VAD-related increases of the proliferation of these cells, regression of their differentiation stage, and by using DNA microarray technology to further define the consequences of VAD on their transcriptome.
Sertoli cells are somatic cells located in the seminiferous tubules. They are essential for an adequate and protected environment for germ cell development within the seminiferous tubules. We observed that the Sertoli cells from VAD animals lost their ability to adhere to culture dishes, suggesting a disruption of the extracellular matrix and/or cell junctions. Using real-time PCR, we noticed a VAD-induced decrease of the expression level of GAP-43, a protein involved in the formation of the GAP junctions. The literature suggests an involvement of GAP-43 and thyroid hormones in Sertoli cell proliferation and differentiation. Hypothyroidism, associated with a decrease of GAP-43 expression, has been linked to an increase in Sertoli cells numbers and regression to a less differentiated stage. We first compared the number of Sertoli cells in the VAD animals with those in control animals, using vimentin, a cytoplasmic marker of these cells, and immunohistochemistry; we found a VAD-related increase of the number of vimentin-positive cells, results that we confirmed by a Western blot analyses, suggesting an increased number of Sertoli cells in the VAD animals.
We selected some markers for real-time PCR analysis to analyze the proliferation/differentiation status of Sertoli cells: Cyclin D2, a marker of proliferation; AMH, a marker of undifferentiation; and keratin 18, a marker of differentiation. We are currently isolating pure populations of Sertoli cells from control and treated animals, using a combination of the STAPUT method and flow cytometry, and we will use the markers described above to compare the proliferation/differentiation status of these cells.
We also plan to use microarray analysis to compare the expression profile of the Sertoli cells from VAD and control animals. In parallel, we are studying the effect of VAD on Leydig cells, interstitial cells that are adjacent to the seminiferous tubules and that regulate spermatogenesis by producing testosterone. Our initial results show that VAD causes a decline in the serum concentration of testosterone.
Characterization of functional long non-coding RNAs in male germ cell development
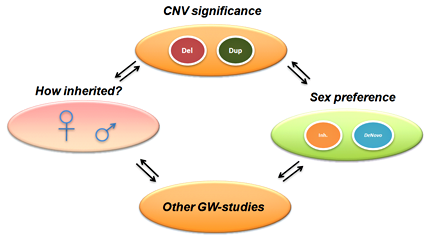
Figure 3. CNN overview, incorporating multi-dimensional biological data in CNV analysis
CNN, CNV-centric node network; CNV, copy number variation; GW, genome-wide. (click image to enlarge)
Spermatogenesis is a key process in mammalian reproduction. This highly ordered process requires precise and well-controlled programs governed by dynamic patterns of gene expression. Although key genes in male germ cell development have been identified, the biological mechanisms and transcripts that govern the programs of spermatogonial stem cell renewal and germ cell differentiation during spermatogenesis or fertilization remain largely unknown.
Other than protein-coding genes, the mammalian genome produces thousands of non-coding RNAs (ncRNAs) of unknown function. Often considered "junk," these non-protein-coding portions of the genome were recently shown to have various biological functions. Long ncRNAs (lncRNAs) are generally classified as ncRNAs longer than 200bp. They function in developmental regulation, such as mouse ESCs pluripotency and differentiation. We hypothesized that they may be indispensible during male germ cell development. This project identified male germ cell–specific lncRNAs candidates and their regulatory functions during male gamete differentiation and development. The objectives include:
- Identify lncRNAs expressed in spermatogenesis from established male germ cell Serial Analysis of Gene Expression (SAGE) libraries and tiling microarray expression data in spermatogonia (Spga), spermatoctes (Spcy), and spermatids (Sptd).
- Validate expression profile during major male germ cell stages.
- Explore their functional importance.
Based on our male germ cell database GermSAGE, we identified 50, 35, and 24 potential lncRNA candidates in Spga, Spcy, and round Sptd, respectively. We classified the lncRNAs based on various genomic location features, including promoter, intronic, intergenic and 3'UTR. The top ten candidates were selected from each stage for subsequent analysis. Our preliminary data suggested that two Spga-related lncRNAs were involved in regulating cell differentiation and were under the control of retinoic acid through its binding sites in the promoter region. Both candidates also demonstrated significant decrease in expression in Spga in VAD animals. Some ncRNAs exhibited more than a thousand-fold decrease compared with control testis. In vitro analysis by a p19 cell differentiation model suggested that other components in the retinoic acid signaling pathway were affected by the Spga lncRNAs (Figure 2). Overexpression of Spga lncRNAs led to repression of components associated with reduced cellular differentiation. The results suggest that long ncRNA may play an active role in spermatogonial stem cell differentiation during male germ cell development.
Biological function of N-terminal acetyltransferase and its gene expression regulation
Protein N-terminal acetylation is mediated in mammalian cells by N-terminal acetyltransferase (NAT). The major form of NAT in human and mouse cells is composed of the NARG1 subunit and the catalytic subunit ARD1A, which is indispensable for enzymatic activity. In somatic tissues, the NARG1-ARD1A is the predominant form of NAT. ARD1A is implicated in cancer progression and neuronal dendritic development. In mouse testicular germ cells, however, the ARD1A subunit is replaced by ARD1B after male meiosis. The identification of ARD1B in the testis suggests the importance of protein N-terminal acetylation in the completion of germ cell development. The upstream and downstream targets of ARD1A or ARD1B have not been completely elucidated. To investigate the biological significance of protein N-terminal acetylation, we study the regulatory mechanism of ARD1A and ARD1B gene expression and the biological functions of their products, usind gene knockdown and gene knockout strategies.
We used a human cancer cell culture as a model system in which to study the biological significance of N-terminal acetylation. In contrast to previous reports, we found that ARD1A is the predominant ARD1 isoform expressed in the human cell culture. Similar to its mouse gene ortholog, we discovered that the CpG island located at the promoter region of the human ARD1B gene is subject to DNA methylation and that the expression of human ARD1B is regulated by DNA methylation. We knocked down the expression of ARD1A using siRNA and identified a limited set of genes whose expression levels are affected by the removal of ARD1A gene, suggesting that the biological function mediated by ARD1A is not limited to protein modification. The majority of the affected genes map to uncharacterized genomic loci. Interestingly, the genes upregulated in the absence of ARD1A include a non-coding RNA and genes that are involved in RNA processing and binding. We are now studying the relationship of these genes and transcripts to elucidate the biological role of ARD1A. Meanwhile, we are studying the biological role of ARD1B in testis development using ARD1B knockout mice. Our preliminary data suggest that the effects of ARD1B knockdown manifest themselves late in male germ cell development. Further studies are under way to pinpoint the susceptible stage of development and to identify the protein substrates that are targeted by NAT in male germ cells.
Bioinformatics and genomic studies on male germ cell and gonad development
Serial analysis of gene expression (SAGE) is a powerful genomic tool to detect RNA species based on sequencing approaches. We developed SAGE databases on male germ cells (GermSAGE) and male embryonic gonads (GonadSAGE) that allows rapid profiling on known and novel transcripts.
GermSAGE is a comprehensive expression database on major stages of mouse male germ cell development. It has an average coverage of 150k in each SAGE library. A total of 452,095 tags derived from type A spermatogonia, pachytene spermatocytes, and round spermatids are included.
GonadSAGE is a SAGE database on male gonad development that covers six male mouse embryonic gonad stages, including E10.5, E11.5, E12.5, E13.5, E15.5 and E17.5. The sequence coverage of each SAGE library is beyond 150K, which is the most extensive sequence-based male gonadal transcriptome to date.
GermSAGE and GonadSAGE are freely available at germsage.nichd.nih.gov and gonadsage.nichd.nih.gov respectively.
Bioinformatics of complex disease: development of CNV-centric node network (CNN) models in autistic spectrum disorders
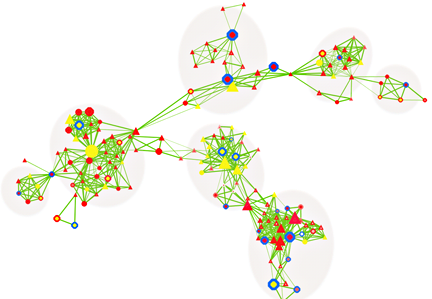
Figure 4. CNV-centric node network (CNN) example in autism spectrum disorders (ASD)
(click image to enlarge)
Autism spectrum disorders (ASD) are characterized by severe deficits in socialization, communication, and repetitive or unusual behaviors. The number of ASD cases is increasing—an important issue impacting our social and medical systems. As a result of multiple confounding factors, the pathogenesis of ASD remains largely unknown. Genetic, epigenetic, and environmental influences as well as dysregulation of immune function have been suggested.
To better understand the underlying mechanisms of ASDs, we attempted to identify key regulatory circuits in ASDs through integrative gene network analysis. In this study, we mainly focused on potential genetic contributors to ASDs through DNA copy number variation (CNV) data from high-throughput genomic assays. CNVs feature small deletions and duplications of chromosomes. Such genetic alterations could be transmitted through inherited (parental or maternal) or non-inherited (de novo) mechanisms. Recent studies have suggested that CNVs could affect gene functions implicated in ASDs.
The majority of CNV data are reported in descriptive formats, usually in form of chromosomal associations and forms of inheritance. Although adequate for reporting purpose, they do not provide insights or point to significant biological implications. We developed a novel approach to integrate multiple biological and functional genomic data with CNV data in form of clustered gene networks (Figure 3). The method is called CNV-centric Node Network (CNN) and is designed is to identify and enhance our understanding of CNV datasets in complex disorders like ASDs. CNN first considers the contributions of CNV data parameters and then compares the significance across similar studies (if any). Significant CNVs were then clustered based on biological evidence (gene ontology or curated biological pathway database). The clusters were also filtered with gene expression data associated with the given CNV to determine if concordant patterns could be identified (Figure 4).
Preliminary CNN analysis highlighted activation of PTEN/TSC1/FMR1 and repression of mTor/PI3K signaling pathways in ASDs, primarily through inherited mechanisms (greater than 70%, Figure 5). CNN provides a novel way to understand the complex genetic mechanisms in ASD and it prioritizes the key CNVs for follow-up studies.
Studies of pediatric patients with metabolic and other genetic disorders
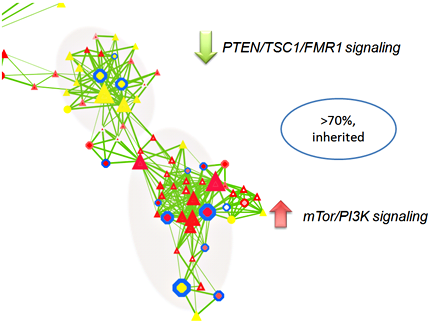
Figure 5. A subset of CNN functional cluster related to PTEN/TSC1/FMR1 and mTor/PI3K signaling in ASD
(click image to enlarge)
Under this protocol, we provide care for patients with a variety of rare genetic disorders. In addition, we supplement and offer an opportunity for training in clinical genetics, dysmorphology, and metabolic genetics at the NICHD and other Institutes of the NIH, and we spearhead the development of new research protocols on particular aspects of diagnosis and care for specific genetic diseases. Evaluations of patients with a broad spectrum of metabolic and genetic conditions are performed; genetic counseling services are offered to patients and their families to assess risk and to give information on preventive measures and testing options. We studied chromosomal and Mendelian disorders of childhood and/or adult onset, congenital anomalies and/or birth defects, dysmorphic syndromes, familial cancer syndromes, multifactorial disorders, and metabolic abnormalities. If not eligible for another NICHD research protocol (specific for a disease or a treatment), patients with genetic/metabolic–related conditions may be evaluated under the auspices of this protocol to advance the clinical skills of physicians participating in NICHD clinical research and training programs and to provide stimuli for new clinical research initiatives. The overall purpose of this protocol is to support the Institute's training and research missions by expanding the spectrum of diseases that can be seen in our clinics and wards. We trained Intramural research Training Awardees (IRTAs), undergraduate and graduate students, medical students, residents, and fellows in the care and management of patients with genetic conditions and their families.
The year 2009-2010 was the 9th year of the protocol. It has been a successful avenue for the training of fellows, residents, and students in the area of metabolic and genetic diseases. This is the 7th year that we have pediatrics residents from Georgetown University Hospital in our protocol. They have rotated in our clinic every month since July, 2003. In addition, in 2007 we began the rotation of all IRTAs in NIH (3 every week). This year, we began a new rotation with the Genetics Fellows and the Genetic Counseling students from NHGRI.
Total number of patients (out- + inpatients until now): 674
Diagnosis information:
| Diagnosis/Problem | # of new patients |
|---|---|
| Menkes disease | 93 |
| Developmental delay/FTT | 119 |
| Family history genetic/metabolic condition | 117 |
| Fetal death/prenatal complications | 5 |
| Breast cancer | 54 |
| Family history of breast cancer | 26 |
| Mitochodria/muscle disease | 22 |
| Dysmorphic features | 32 |
| Failure to thrive | 27 |
| Metabolic disorder | 23 |
| Neurofibromatosis/harmatoses | 13 |
| Chromosomal abnormalities | 13 |
| Short stature/dwarfism | 31 |
| Colobomas | 2 |
| Microcephaly/macro/scapho | 8 |
| Cleft lip/palate | 4 |
| Familial gastro-intestinal tumors (GIST) | 2 |
| Hyperflexibility | 7 |
| Klinefelter syndrome | 6 |
| Café-au-lait spots | 9 |
| Down syndrome | 4 |
| Seizure disorder | 5 |
| Medium chain acyl-CoA dehydrogenase (MCAD) deficiency | 9 |
| Motor neuropathies | 2 |
| Ehlers-Danlos/Marfan syndromes | 5 |
| McCune Albright syndrome | 1 |
| Others | 231 |
Publications
- Lee TL, Chan WY, Rennert OM. Identifying novel long non-coding RNAs in male germ cell development by SAGE. In: Methods in Molecular Biology on Germline Development. Springer Protocol. 2010;in press.
- Lee TL, Rennert OM, Chan WY. Transcriptome landscape in male germ cell development. In: Sperm Chromatin: Biological and Clinical Application in Male Infertility and Assisted Reproduction. Humana Press. 2010;in press.
- Lee TL, Rennert OM. Application of tiling microarray in transcriptome analysis of male germ cell development. In: Methods in Molecular Biology on Germline Development. Springer Protocol. 2010;in press.
- Lee TL, Pang AL, Rennert OM, Chan WY. Genomic landscape of developing male germ cells. Birth Defects Res C Embryo Today. 2009;87(1):43-63.
- Nagrani SR, Levens ED, Baxendale V, Boucheron C, Chan WY, Rennert OM. Methylation patterns of Brahma during spermatogenesis and oogenesis: potential implications. Fertil Steril. 2010;[Epub ahead of print].
- Kokkinaki M, Lee TL, He Z, Jiang J, Golestaneh N, Hofmann MC, Chan WY, Dym M. Age affects gene expression in mouse spermatogonial stem/progenitor cells. Reproduction. 2010;139(6):1011-1020.
- Lee TL, Li Y, Alba D, Vong QP, Wu SM, Baxendale V, Rennert OM, Lau YF, Chan WY. GonadSAGE: a comprehensive SAGE database for transcript discovery on male embryonic gonad development. Bioinformatics. 2010;26(4):585-586.
- Lee TL, Cheung HH, Claus J, Sastry C, Singh S, Vu L, Rennert O, Chan WY. GermSAGE: a comprehensive SAGE database for transcript discovery on male germ cell development. Nucleic Acids Res. 2009;37D:891-897.
- Kokkinaki M, Lee TL, He Z, Jiang J, Golestaneh N, Hofmann MC, Chan WY, Dym M. The molecular signature of spermatogonial stem/progenitor cells in the 6-day-old mouse testis. Biol Reprod. 2009;80(4):707-717.
- Lee TL, Chan WY, Rennert OM. Assessing the safety of nanomaterials by genomic approach could be another alternative. ACS Nano. 2009;22(12):3830.
- Johnston JJ, Sapp JC, Turner JT, Amor D, Aftimos S, Aleck KA, Bocian M, Bodurtha JN, Cox GF, Curry CJ, Day R, Donnai D, Field M, Fujiwara I, Gabbett M, Gal M, Graham JM, Hedera P, Hennekam RC, Hersh JH, Hopkin RJ, Kayserili H, Kidd AM, Kimonis V, Lin AE, Lynch SA, Maisenbacher M, Mansour S, McGaughran J, Mehta L, Murphy H, Raygada M, Robin NH, Rope AF, Rosenbaum KN, Schaefer GB, Shealy A, Smith W, Soller M, Sommer A, Stalker HJ, Steiner B, Stephan MJ, Tilstra D, Tomkins S, Trapane P, Tsai AC, Van Allen MI, Vasudevan PC, Zabel B, Zunich J, Black GC, Biesecker LG. Molecular analysis expands the spectrum of phenotypes associated with GLI3 mutations. Hum Mutat. 2010;31(10):1142-1154.
- Stratakis CA, Tichomirowa MA, Boikos S, Azevedo MF, Lodish M, Martari M, Verma S, Daly AF, Raygada M, Keil MF, Papademetriou J, Drori-Herishanu L, Horvath A, Tsang KM, Nesterova M, Franklin S, Vanbellinghen JF, Bours V, Salvatori R, Beckers A. The role of germline AIP, MEN1, PRKAR1A, CDKN1B and CDKN2C mutations in causing pituitary adenomas in a large cohort of children, adolescents, and patients with genetic syndromes. Clin Genet. 2010;[Epub ahead of print].
- Raygada M, Arthur DC, Wayne AS, Rennert OM, Toretsky JA, Stratakis CA. Juvenile xanthogranuloma in a child with previously unsuspected neurofibromatosis type 1 and juvenile myelomonocytic leukemia. Pediatr Blood Cancer. 2010;54(1):173-175.
Collaborators
- Joan Han, MD, Program on Developmental Endocrinology and Genetics, NICHD, Bethesda, MD
- Constantine Stratakis, MD, D(med)Sci, Program on Developmental Endocrinology and Genetics, NICHD, Bethesda, MD
- Yan A. Su, MD, PhD, George Washington University, Washington, DC
Contact
For more information, email rennerto@mail.nih.gov.