

You are here: Home > Section on Clinical and Developmental Genomics
Genetic and Genomic Studies in Normal Development and Diseases

- Owen M. Rennert, MD, Head, Section on Clinical Genomics
- Tin Lap Lee, PhD, Staff Scientist
- Lap-Yin Pang, PhD, Biologist
- Margarita Raygada, MS, PhD, Genetic Counselor
- Catherine Boucheron, PhD, Postdoctoral Fellow
- Hoi Hung Cheung, BS, MS, Visiting Fellow
- Wai Yee Chan, PhD, Volunteer
- Shao Ming Wu, PhD, Volunteer
- Xiaozhuo Liu, MS, PhD, Predoctoral Visiting Fellow
- Vanessa Baxendale, BS, Laboratory Manager
- Lucile Thouennon, Registered Laboratory Animal Technician (Contractor)
- Cathering A. Edmonson, BS, Postbaccalaureate Intramural Research Training Award Fellow
- Alexandra C. Title, BS, Postbaccalaureate Intramural Research Training Award Fellow
- Mark Ziats, BS, Postbaccalaureate Intramural Research Training Award Fellow
SAGE spermatogenesis database
We capitalized on our previous publication of the SAGE Spermatogenesis Database to further explore male germ cell transcriptome complexity by serial analysis of gene expression (SAGE) and to profile the expression signature of the major stages of spermatogenesis, including type A spermatogonia (Spga), pachytene spermatocytes (Spcy), and round spermatids (Sptd). The resultant genomic data led to discovery of a large number of transcripts, including novel, alternative splicing variants, antisense, as well as transcriptional regulatory networks. Preliminary analysis suggested that more than half of the transcriptome was not annotated. The transcriptional architecture implies that most genomic regions serve many functions. Although a large proportion of human transcription occurs outside the boundaries of known genes, the functional significance of the transcripts remains unknown. Based on our male germ cell SAGE libraries, we mapped a total of 29,654 SAGE tags to at least one germ cell stage, and identified several alternative splicing variants spliced from the same gene. 73 genes exhibited different 3′ alternative splicing in all the three stages. We identified novel variants involved in developmental and transcriptional control, such as heat shock protein 4 (Hspa4), H3 histone family 3B (H3f3b), and ubiquitin protein ligase E3A (Ube3a). Our research highlighted that noncoding RNAs (ncRNA) can have a wide range of functions and may be subdivided into different classes by size or function. The SAGE and tiling microarray data demonstrated more than 80% of transcript species were not annotated by current gene annotations. Long noncoding RNA (lncRNA) candidates and their functional importance in male germ cell development remain largely unknown.
Testicular germ cell tumor (TGCT)
Testicular germ cell tumor (TGCT) is a common malignancy in young males. Sensitive tumor markers, accurate prognostic classification and high 5-year survival (dependent upon chemotherapy) make it a good model to study cancer therapy. Molecular modification of DNA methylation is a frequent epigenetic alteration associated with cancer progression. Though aberrant DNA methylation is implicated in the pathophysiology of many cancers, only a limited number of genes are epigenetically changed in TGCT. We used methylated DNA immunoprecipitation (MeDIP) and whole-genome tiling arrays to analyze differentially methylated regions (DMR) in testicular embryonal carcinoma NT2 cells (a non-seminomatous TGCT cell line). We identified a total of 35,208 DMRs. However, only a small number of DMRs mapped to promoters. Microarray gene expression analysis yielded a group of differentially expressed genes regulated by DNA methylation. Several candidate genes, including APOLD1, PCDH10 and RGAG1, were dysregulated in TGCT patient samples. Surprisingly, the APOLD1 gene was previously mapped to the 12p13.1 TGCT susceptibility locus, suggesting it may be important in TGCT pathogenesis. We also observed aberrant methylation in the loci of several miRNAs (miR-199a, miR-124a, and miR-184) and snoRNAs (HBII-240, ACA33, and ACA8). Our study was the first application of MeDIP-chip for identifying epigenetically regulated genes and non-coding RNAs in TGCT. We documented the potential function of intergenic and intronic DMRs in the regulation of ncRNAs.
Subsequently we focused on the miR-199a, an important miRNA, documented by others, to be associated with the progression and prognosis of gastric and ovarian cancer. Located at chromosome 1q24.3, miR-199a is transcribed as antisense of dynamin 3. We found that hypermethylation of this region correlated with repression of miR-199a-5p/3p and tumor malignancy. Re-expression of miR-199a in testicular cancer cells led to suppression of cell growth, cancer migration, invasion, and metastasis. The miR-199a-5p, one of two mature miRNA species derived from miR-199a, was associated with tumor malignancy. We further identified the embryonal carcinoma antigen podocalyxin-like protein 1 (PODXL), an anti-adhesive protein expressed in aggressive tumors, as a target of miR-199a-5p. We documented that PODXL is over-expressed in malignant testicular tumor, and its depletion resulted in suppression of cancer invasion. The inverse relationship between PODXL and miR-199a-5p expression suggests that PODXL is a downstream effector mediating the action of miR199a-5p. The results identified DNA methylation, miR-199a dysregulation, and PODXL as critical factors in tumor malignancy.
We extended our study to epigenetic profiling of a group of non-seminomatous TGCT, testicular embryonal carcinoma (EC). We profiled DNA methylation for six ECs of different clinical stages. Genome-wide methylation analysis revealed a distinct methylation signature between normal tissue and tumors. We identified DMRs specific for metastatic ECsd, the majority of which are hypermethylation. Mapping of hypermethylated DMRs to Refseq identified only 39 DMRs located in gene promoters. The majority of DMRs (about 97%) were located in gene bodies or non-genic regions, consistent with our previous finding in testicular cancer cell lines. Interestingly, we found several sex-linked genes that were hypermethylated, including the X-linked genes STAG2, SPANXD/SPANXE, MIR1184, the Y-linked genes RBMY1A1/RBMY1B/RBMY1D, BTBD2, ZNF699, and FAM197Y2P. We selected nine genes for qPCR analysis. Among the genes analyzed, expression of AGPAT3, SUCLG2, RBMY1A, SPANXD, RNF168, USP13, and FAM197Y2P were significantly reduced in ECs. One of the Y-linked genes, RBMY1A, has been reported to be regulated by DNA methylation in prostate cancer. The role of RBMY1A in male germ cell tumor is not well understood. Immunohistochemical analysis of normal testis, EC, and seminoma tissues indicated down-regulation of this protein in testicular tumors. Expression of RBMY1A was restricted to male germ cells and disappeared in ECs, and seminomas. Our genome-wide analysis identified methylation changes in several previously unknown genes for testicular ECs, possibly providing insight into the crosstalk between normal germ cell development and carcinogenesis.
Bioinformatics and genomic studies on male germ cell and gonad development
We developed SAGE databases on male germ cells (GermSAGE) and male embryonic gonads (GonadSAGE) that allows rapid profiling of known and novel transcripts. The databases permit extracting cellular dynamics contained in the dataset, such as transcriptional regulation through over-representation or co-expression of promoter sequence elements and gene interaction networks at a particular germ cell stage. GermSAGE and GonadSAGE are freely available at germsage.nichd.nih.gov and gonadsage.nichd.nih.gov respectively and are useful to investigators throughout the world.
Autism spectrum disorders
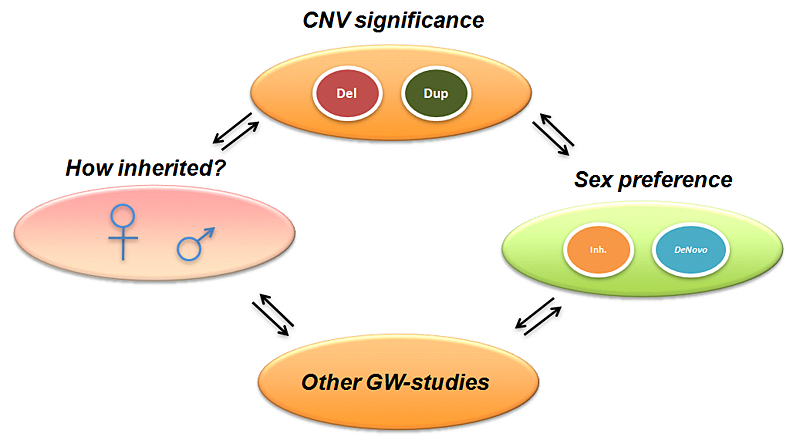
Click image to enlarge.
Figure 1. CNN overview, incorporating multidimensional biological data in CNV analysis
CNN, CNV-centric node network; CNV, copy number variation; GW, genome-wide
Autism spectrum disorders (ASD) are characterized by severe deficits in socialization, communication, and repetitive or unusual behaviors. The prevalence of ASD cases is increasing, and it is an important public health issue. Owing to serious multiple confounding factors, the pathogenesis of ASD remains largely unknown. Genetic and epigenetic factors along with dysregulation of immune function and environmental influences have been suggested. The ASDs represent a clinically heterogeneous set of conditions with strong hereditary components. Despite substantial efforts to uncover the genetic basis of ASDs, the genomic etiology appears complex, and a clear understanding of the molecular mechanisms underlying autism remains elusive.
To better understand the underlying mechanisms of ASDs, we attempted to identify key regulatory circuits in ASDs through integrative gene network analysis. We mainly focused on potential genetic contributions of ASDs through DNA copy number variation (CNV) data from high-throughput genomic assays. CNVs identify small deletions and duplications of chromosomes. Such genetic alterations could be transmitted through inherited (paternal or maternal) or non-inherited (de novo) mechanisms. Recent studies suggested that CNVs could affect gene functions that are implicated in ASDs. We developed a novel approach to integrate diverse biological evidence with functional genomic data to the CNV data in the form of clustered gene networks.
Figure 1 displays the method called CNV-centric Node Network (CNN). The concept underlying CNN is to identify and enhance the understandings of CNV datasets in complex disorders like ASDs. CNN first considers the contributions of CNV data parameters and compares their significance across similar studies (if any). Significant CNVs were then clustered based on biological evidence (gene ontology or curated biological pathway database). The clusters were also filtered with gene expression data associated with the given CNV to determine whether a concordant pattern could be identified.
To integrate the genomic heterogeneity underlying the complex etiologies of common neurodevelopmental disorders, we analyzed expression of all implicated genes in autism, schizophrenia, and epilepsy using next-generation transcriptome sequencing in the developing human brain. To our knowledge, no study has yet attempted to derive molecular pathways underlying ASD by investigating all implicated genes together. To do so, we first described gene ontology, canonical pathways, and interactome networks for all genes implicated in ASD. We are the first to report the gene expression profile of all ASD-implicated genes in the unaffected developing human brain. By implementing a novel computational approach, we identified a subset of highly expressed ASD-candidate genes from which interactome networks were derived. Strikingly, immune signaling through NFκB, Tnf, and Jnk was central to ASD networks at multiple levels of our analysis, and cell type–specific expression suggested glia—in addition to neurons—deserve consideration. This work will serve as an important resource in autism research and provides integrated genomic evidence for a role of the immune system in ASD.
Clinical Training at NICHD
Our Clinical protocol 02-CH-0023 provides care for patients with a variety of genetic and metabolic disorders; comprehensive evaluations are offered to patients with suspected or diagnosed genetic conditions. In addition, we supplement and offer an additional opportunity for training in clinical genetics, dysmorphology, and metabolic genetics at the NICHD and other Institutes of the NIH. The year 2010–2011 was the 10th year of the protocol; we have trained fellows, medical students, residents, genetic counseling students, Intramural Research Training Awardees (IRTA), and MD/PhD students across the NIH and other medical centers. This is the eighth year that we have pediatric residents from Georgetown University Hospital in our protocol. In addition, in 2007 we began the rotation of all IRTAs in NIH. Last year we began a new rotation with the Genetics Fellows from NHGRI and have now added Genetic Counseling students from NHGRI, and begun inpatients rounds with MD/PhD students at NIH. Our training allows trainees to acquire broad knowledge of the heterogeneity, variability, and natural history of genetic/metabolic disorders in order to make decisions about patient care and counseling on preventive measures. Our protocol has also spearheaded the development of new research protocols on particular aspects of diagnosis and care for specific genetic diseases, among which is the new protocol for analysis of speech and language development in developmentally delayed children. In addition, genetic counseling services are offered to patients and their families to assess risk and give information on preventive measures and testing options. The counseling services are also offered to patients in other clinical protocols as part of our training function. Chromosomal and Mendelian disorders of childhood and/or adult onset are studied as are congenital anomalies and/or birth defects, dysmorphic syndromes, familial cancer syndromes, multifactorial disorders, and metabolic abnormalities. A major purpose of this protocol is to recruit a diverse population of subjects and/or samples with a known or suspected genetic/metabolic disorder in order to provide NICHD investigators and trainees with hands-on experience related to diagnosis, management, follow-up, treatment, and genetic counseling. The protocol also provides an opportunity to evaluate patients with genetic and metabolic disorders that may not be eligible for an existing research protocol; often it is not possible to determine protocol eligibility without prospective evaluation conducted at the NIH. Patients and/or family members with genetic disorders may offer their DNA for storage and/or testing. The overall purpose of this protocol is to support our Institute's training and research missions by expanding the spectrum of diseases that can be seen in our clinics and wards.
Publications
- Lee TL, Cheung HH, Claus J, Sastry C, Singh S, Vu L, Rennert O, Chan WY. GermSAGE: a comprehensive SAGE database for transcript discovery on male germ cell development. Nucleic Acids Res 2009;37D:891-897.
- Cheung HH, Lee TL, Davis AJ, Taft DH, Rennert OM, Chan WY. Genome-wide DNA methylation profiling reveals novel epigenetically regulated genes and non-coding RNAs in human testicular cancer. Br J Cancer 2010;102:419-427.
- Cheung HH, Davis AJ, Lee TL, Pang AL, Nagrani S, Rennert OM, Chan WY. Methylation of an intronic region regulates miR-199a in testicular tumor malignancy. Oncogene 2011;30:3404-3415.
- Ziats, M, Rennert OM. Expression Profiling of Autism Candidate Genes during Human Brain Development Implicates Central Immune Signaling Pathways. Plos One 2011;6(9):e24691.
- Pang AL, Clark J, Chan WY, Rennert OM. Expression of human NAA11 (ARD1B) gene is tissue-specific and is regulated by DNA methylation. Epigenetics 2011;6:[Epub ahead of print].
Collaborators
- Joan Han, MD, Program in Developmental Endocrinology and Genetics, NICHD, Bethesda, MD
- Lynne Holtzclaw, BS, Imaging Core, NICHD, Bethesda, MD
- James Russell, PhD, Imaging Core, NICHD, Bethesda, MD
- Vincent Schram, PhD, Imaging Core, NICHD, Bethesda, MD
- Beth Solomon, MS, Rehabilitation Department, NIH Clinical Center, Bethesda, MD
- Constantine Stratakis, MD, D(med)Sci, Program in Developmental Endocrinology and Genetics, NICHD, Bethesda, MD
- Susan Swedo, MD, Pediatrics & Developmental Neuroscience Branch, NIMH, Bethesda, MD
- Audrey Thrum, PhD, Pediatrics & Developmental Neuroscience Branch, NIMH, Bethesda, MD
Contact
For more information, email rennerto@mail.nih.gov or visit scg.nichd.nih.gov.