
Protein Sorting in the Endomembrane System
- Juan S. Bonifacino,
PhD, Head, Section on Intracellular Protein Trafficking - Raffaella De Pace, PhD, Staff Scientist
- Adriana Golding, PhD, Research Fellow
- William Huffman, MS, Technician
- Xiaolin Zhu, Technician
- Nireekshit Addanki Tirumala, PhD, Visiting Fellow
- Felipe Del Valle Batalla, PhD, Visiting Fellow
- Saikat Ghosh, PhD, Visiting Fellow
- Zhaoxing Ran, PhD, Visiting Fellow
- Ganesh Shelke, PhD, Visiting Fellow
- Surbhi Verma, PhD, Visiting Fellow
- Jennifer M. Kunselman, PhD, Intramural Research Training Award Fellow
- Rafael Mattera, PhD, Special Volunteer

Our laboratory studies the molecular mechanisms underlying the sorting of transmembrane proteins (known as cargo) to various compartments within the endomembrane system of eukaryotic cells. The system comprises an array of membrane-enclosed organelles, including the endoplasmic reticulum (ER), the Golgi apparatus, the trans-Golgi network (TGN), endosomes, lysosomes, lysosome-related organelles (LROs, e.g., melanosomes, cytotoxic granules), and various domains of the plasma membrane in polarized cells such as epithelial cells and neurons. The transport of cargo between these compartments is mediated by vesicular or tubular carriers that bud from a donor compartment, translocate through the cytoplasm, and fuse with an acceptor compartment. We study the molecular machineries that mediate these processes in the context of different intracellular transport pathways, including endocytosis, recycling from endosomes to the plasma membrane, retrograde transport from endosomes to the TGN, biogenesis of lysosomes and LROs, autophagy, and polarized sorting in epithelial cells and neurons. Our fundamental research serves as a basis for explaining the pathogenetic mechanisms of protein and organelle transport disorders, including the pigmentation and bleeding disorder Hermansky-Pudlak syndrome (HPS), hereditary spastic paraplegias (HSPs), and other neurodevelopmental disorders.
Figure 1. Schematic representation of the endomembrane system of eukaryotic cells showing the localization of coats involved in protein sorting
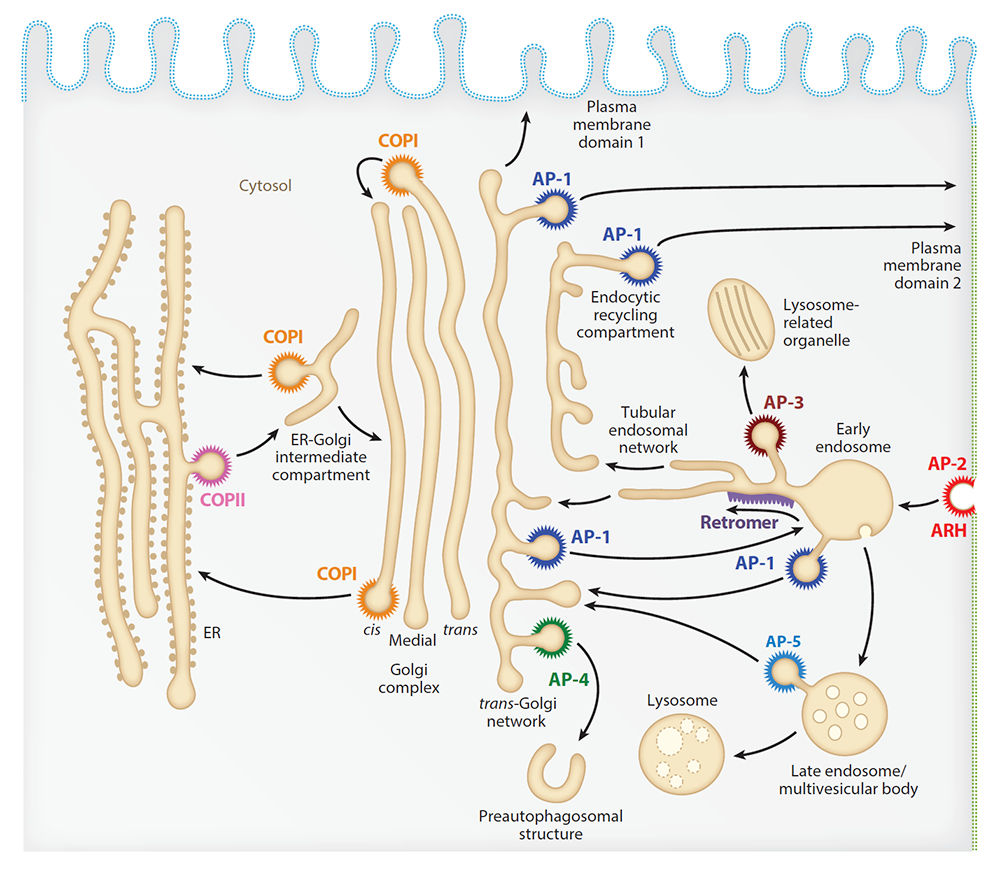
Figure 1. Schematic representation of the endomembrane system of eukaryotic cells showing the localization of coats involved in protein sorting
Messenger RNA transport on lysosomal vesicles maintains axonal mitochondrial homeostasis and prevents axonal degeneration.
In neurons, RNA granules are transported along the axon for local translation away from the soma. Recent studies indicate that some of this transport involves the hitchhiking of RNA granules on lysosome-related vesicles. This past year, to identify a subset of axonal mRNAs that depend on lysosome-related vesicles for transport, we leveraged the ability to prevent transport of these vesicles into the axon by knocking out the lysosome-kinesin adaptor BLOC-one-related complex (BORC), which we discovered in previous research. We found that BORC knockout causes depletion of a large group of axonal mRNAs mainly encoding ribosomal and mitochondrial/oxidative phosphorylation proteins. The depletion results in mitochondrial defects and eventually leads to axonal degeneration in human induced pluripotent stem cell (iPSC)–derived and mouse neurons. Pathway analyses of the depleted mRNAs revealed a mechanistic connection between BORC deficiency and common neurodegenerative disorders. The findings demonstrate that mRNA transport on lysosome-related vesicles is critical for the maintenance of axonal homeostasis and that its failure causes axonal degeneration.
Biallelic BORCS8 variants cause an infantile-onset neurodegenerative disorder with altered lysosome dynamics.
BORC is a multiprotein complex composed of eight subunits, named BORCS1 through BORCS8. It associates with the cytosolic face of lysosomes, recruiting the small GTPase ARL8 and kinesin-1 and kinesin-3 microtubule motors to promote anterograde transport of lysosomes toward the peripheral cytoplasm in non-neuronal cells and the distal axon in neurons. The physiological and pathological importance of BORC in humans remains unclear. This past year, in collaboration with Reza Maroofian, Emily Farrow, Shunmoogum Patten, and colleagues, we identified compound heterozygous and homozygous variants in BORCS8 in children with a severe early-infantile neurodegenerative disorder. The children exhibited global developmental delay, severe-to-profound intellectual disability, hypotonia, limb spasticity, muscle wasting, dysmorphic facies, optic atrophy, leuko-axonopathy with hypomyelination, and neurodegenerative features with prevalent supratentorial involvement. Cellular studies using a heterologous transfection system showed that the BORCS8 variants exhibited reduced expression, less assembly with other BORC subunits, and/or lower ability to drive lysosome distribution toward the cell periphery. Therefore, all BORCS8 variants identified in this study are likely pathogenic, loss-of-function alleles. Knockout of the orthologous borcs8 in zebrafish caused decreased brain and eye size, neuromuscular anomalies, and impaired locomotion, recapitulating key traits of the human disease. The findings identified BORCS8 as a novel genetic locus for an early-infantile neurodegenerative disorder and highlighted the critical importance of BORC and lysosome dynamics for the development and function of the central nervous system.
AP2A2 mutation and defective endocytosis in a Malian family with hereditary spastic paraplegia
Hereditary spastic paraplegia (HSP) is a group of neurogenetic disorders characterized by progressive lower extremity spasticity. This past year, we participated in a study with Christopher Grunseich, Kenneth Fischbeck, Guida Landouré, and colleagues that identified children from a Malian family with symptoms consistent with childhood-onset complicated HSP. Neurological evaluation revealed lower limb weakness, spasticity, dysarthria, seizures, and intellectual disability. Brain MRI showed corpus-callosum thinning with cortical and spinal cord atrophy, and EEG detected a slow background in the index patient. Whole-exome sequencing identified a homozygous missense variant in the adaptor protein complex 2 alpha-2 subunit (AP2A2) gene. Cellular studies showed reduced levels of AP2A2, of endocytosis of the transferrin receptor (TfR), and of axon initial segment length in patient-derived neurons. Additionally, the AP2A2 variants exhibited defective binding to accessory proteins. The findings identified AP2A2 as a novel genetic entity associated with HSP, expanding our understanding of the disease and improving its genetic diagnosis.
Revising pathogenesis of AP1S1–related MEDNIK syndrome: a missense variant in the AP1S1 gene as a causal genetic lesion
MEDNIK syndrome is a rare autosomal recessive disorder characterized by mental retardation, enteropathy, deafness, peripheral neuropathy, ichthyosis, and keratoderma, which are caused by variants in the AP1S1 gene. The gene encodes sigma1A, a subunit of the adaptor protein complex 1 (AP-1), which plays a key role in intracellular protein trafficking. Previously, AP1S1 nonsense, frameshift, and splice-site variants were identified in MEDNIK patients, predicted to produce truncated sigma1A proteins, leading to AP-1 dysfunction. Recently, an AP1S1 c.269 T→C missense variant was reported in patients with severe enteropathy but without other MEDNIK features, suggesting a novel, non-syndromic form of congenital diarrhea caused by this missense variant. This past year, in collaboration with the laboratory of Karolina Skvarova Kramarzova, we reported the characterization of two patients with the same missense variant, who, in contrast to previous cases, exhibit complete MEDNIK syndrome. The findings revise the clinical spectrum of AP1S1 variants, indicating that all pathogenic AP1S1 variants can cause MEDNIK syndrome. Additionally, our functional analyses demonstrate the impact of the missense variant on sigma1A function, enhancing our understanding of MEDNIK syndrome’s molecular pathogenesis.
Contribution to the development of intrathecal AAV9/AP4M1 gene therapy for hereditary spastic paraplegia 50 caused by mutations in the mu4 subunit of AP-4
We contributed to the development of gene therapy for hereditary spastic paraplegia 50 (SPG50) in collaboration with Xin Chen, Steven Gray, and other colleagues. SPG50 is a rare childhood-onset neurological disorder caused by mutations in the AP4M1 gene. Our laboratory demonstrated that infection of skin fibroblasts from patients with AP4M1 mutations with an AAV2–AP4M1 vector rescued the assembly of the AP-4 complex and the export of ATG9A from the trans-Golgi network. Our collaborators then showed that intrathecal injection of a similar AAV9–AP4M1 vector had an acceptable safety profile in mice, rats, and non-human primates, and resulted in partial correction of phenotypic defects in Ap4m1–KO mice, preclinical results that support an investigational gene transfer clinical trial to treat SPG50.
Architecture of the ESCPE-1 membrane coat: unveiling a key process of endosomal sorting
Intracellular recycling of membrane proteins plays a crucial role in re-using receptors, ion channels, and transporters. A critical player in this recycling machinery is the endosomal sorting complex for promoting exit 1 (ESCPE-1), responsible for rescuing transmembrane proteins from the endolysosomal pathway and transporting them to the trans-Golgi network and the plasma membrane. We collaborated with the laboratories of Aitor Hierro and Daniel Castaño-Díez to conduct biochemical, structural, and functional analyses into the organization of ESCPE-1, revealing that the complex forms a coat with a single-layer architecture. Furthermore, we found that synergistic interactions between ESCPE-1 protomers, phosphoinositides, and cargo molecules lead to the global arrangement of amphipathic helices, driving the formation of recycling tubules.
Endolysosome fusion attenuates exosome secretion.
The eight-subunit complex BORC plays a crucial role in recruiting the small GTPase ARL8, kinesin motor proteins, and the tethering factor HOPS to late endosomes and lysosomes. This past year, we reported that the BORC–ARL8–HOPS axis is responsible for regulating exosome secretion. Exosomes are small vesicles that cells release to dispose of undegraded materials and they facilitate intercellular communication. A major source of exosomes is intraluminal vesicles within multi-vesicular endosomes, which can undergo either exocytic fusion with the plasma membrane or fusion with lysosomes. However, the factors that determine these alternative fates were previously unknown. Our findings showed that disrupting the BORC–ARL8–HOPS axis impairs endolysosomal fusion, preventing the delivery of intraluminal vesicles to lysosomes, thus increasing exosome secretion. Additionally, our findings suggested that targeting the BORC–ARL8–HOPS pathway may be a promising strategy to enhance exosome yields for biotechnology applications.
Publications
- Messenger RNA transport on lysosomal vesicles maintains axonal mitochondrial homeostasis and prevents axonal degeneration. Nat Neurosci 2024 27:1087–1102
- Biallelic BORCS8 variants cause an infantile-onset neurodegenerative disorder with altered lysosome dynamics. Brain 2024 147:1751–1767
- Revising pathogenesis of AP1S1-related MEDNIK syndrome: a missense variant in the AP1S1 gene as a causal genetic lesion. J Mol Med 2024 102:1343–1353
- Angiogenesis is limited by LIC1-mediated lysosomal trafficking. Angiogenesis 2024 27:943–962
- SLC26A4-AP-2 mu2 interaction regulates SLC26A4 plasma membrane abundance in the endolymphatic sac. Sci Adv 2024 10:eadm8663
- AP2A2 mutation and defective endocytosis in a Malian family with hereditary spastic paraplegia. Neurobiol Dis 2024 198:106537
Collaborators
- Daniel Castaño-Díez, BSc, University of Basel, Basel, Switzerland
- Xin Chen, PhD, University of Texas Southwestern Medical Center, Dallas, TX
- Ryan K. Dale, MS, PhD, Bioinformatics and Scientific Programming Core, NICHD, Bethesda, MD
- Steven Gray, PhD, University of Texas Southwestern Medical Center, Dallas, TX
- Emily G. Farrow, PhD, CGC, FAC, University of Missouri-Kansas City School of Medicine, Kansas City, MO
- Kenneth Fischbeck, MD, Neurogenetics Branch, NINDS, Bethesda, MD
- Christopher Grunseich, MD, PhD, Inherited Neuromuscular Diseases Unit, NINDS, Bethesda, MD
- Aitor Hierro, PhD, CIC bioGUNE, Bilbao, Spain
- Guida Landouré, PhD, University of Science, Technique and Technology of Bamako, Mali
- Reza Maroofian, PhD, University College London, London, United Kingdom
- Maria P. Marzolo, PhD, Pontifical Catholic University, Santiago, Chile
- Nicole Y. Morgan, PhD, Center for Biomedical Engineering Technology Acceleration (BETA), NIBIB, Bethesda, MD
- Shunmoogum A. Patten, PhD, INRS-Institut Armand Frappier, Laval, Canada
- Isabelle Roux, PhD, Section on Human Genetics, NIDCD, Bethesda, MD
- Karolina Skvarova Kramarzova, MD, Charles University and University Hospital Motol, Prague, Czech Republic
- Amber Stratman, PhD, WashU Medicine, St. Louis, MO
- Michael E. Ward, MD, PhD, Inherited Neurodegenerative Diseases Unit, NINDS, Bethesda, MD
- Brant Weinstein, PhD, Section on Vertebrate Organogenesis, NICHD, Bethesda, MD
Contact
For more information, email juan.bonifacino@nih.gov or visit https://www.nichd.nih.gov/research/atNICHD/Investigators/bonifacino.