



You are here: Home > Unit on Pediatric Genetics
Translational Research on Mammalian Copper Transport Disorders

- Stephen G. Kaler, MD, MPH, Head, Unit on Pediatric Genetics*
- Anthony Donsante, PhD, Postdoctoral Fellow
- Julia D. Hicks, BS, Postbaccalaureate Fellow
- Jingrong Tang, MD, Biologist
* As of December 1, 2019, Dr. Kaler is now at Nationwide Children’s Hospital.
The Unit on Pediatric Genetics conducts translational research that focuses on the diagnosis, treatment, and understanding of copper metabolism disorders, a recently expanded collection of human diseases that impact various components of the central and peripheral nervous systems. Acquired, as well as inherited, forms of abnormal copper homeostasis are implicated in neurologic dysfunction (Desai and Kaler, 2008). Menkes disease (MD), a prototypical genetic syndrome of defective copper transport, is characterized by low brain copper levels, infantile neurodegeneration, and premature death. The condition is caused by mutations in ATP7A, an X-linked copper transporter gene. The occipital horn syndrome is a milder allelic variant, associated with leaky ATP7A splice junction mutations, in which autonomic dysfunction predominates. Recently, we identified several families with motor neuron disease resembling Charcot-Marie-Tooth disease and amyotrophic lateral sclerosis that are caused by novel missense mutations in the carboxyl half of ATP7A. These conditions are the consequence of mild loss-of-function defects that appear to affect intracellular trafficking of ATP7A. Thus, the phenotypic spectrum of ATP7A mutations has expanded; discovery of these new allelic variants indicates a previously unappreciated role for ATP7A in motor neuron maintenance and function.
Menkes disease
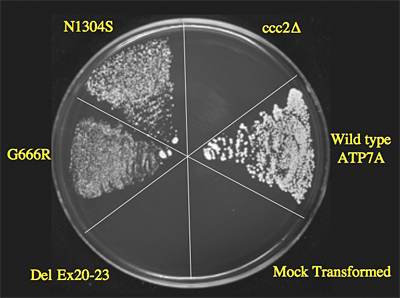
Figure 1. Yeast complementation assay predicts responsivity to early treatment in Menkes disease.
The plating pattern (clock-wise from 12 o’clock) includes the yeast copper transport mutant ccc2∆; ccc2∆ transformed with the normal (wild-type; WT) ATP7A allele; ccc2∆ mock-transformed with an empty vector; ccc2∆ transformed with a mutant ATP7A allele harboring deletion of exons 20-23; ccc2∆ transformed with mutant ATP7A allele G666R; and ccc2∆ transformed with the mutant ATP7A allele N1304S (associated with typical Occipital Horn syndrome, a mild variant of Menkes disease, and used as a positive control). Yeast strains were plated on four different media: normal YPD; synthetic yeast nitrogen base (YNB) supplemented with copper (“copper-sufficient” medium); YNB supplemented with iron (“iron-sufficient” medium); and copper/iron–limited YNB. All strains grew on YPD, copper-sufficient and iron-sufficient media (data not shown), whereas only the WT, G666R, and N1304S show growth on copper/iron–limited media, indicating the copper transport activity by these alleles. The WT allele shows the most robust growth. The del exon 20-23 allele failed to complement the knockout strain, indicating no residual copper transport activity. Two patients with G666R achieve fair to excellent neurodevelopmental outcomes, in contrast to one patient with del ex20-23, whose outcome was poor despite early identification and treatment. From Kaler et al., N Engl J Med.
Current treatment for MD is limited to subcutaneous copper injections and is effective in patients with mutant alleles that do not completely abrogate ATP7A function (Figure 1) who are identified near birth (Kaler et al, 2008 and 2009). For patients with complete loss-of-function mutations, however, copper delivered peripherally does not cross the blood-brain barrier efficiently, and alternative treatment approaches are needed. In our recent exploration of treatment alternatives, we rescued a mouse model of severe Menkes disease using combination brain-directed therapies: a recombinant adeno-associated virus serotype 5 (AAV5) vector expressing a reduced-size human ATP7A, plus copper (Session #42/Abstract #143, Annual Meeting of The American Society of Human Genetics; http://www.ashg.org/2009meeting/). Neither treatment alone was effective, but the combination therapy dramatically shifted the Kaplan-Meier survival curve, with 5 of 16 (31%) combination-treated mutants surviving beyond 110 days of age (p<0.0002) (Figure 2). Identification of the mechanisms underlying this pronounced synergistic effect will help illuminate the normal processes of copper transport in the brain and the role of ATP7A in neuronal and neuroglial cells. In addition, this advance in gene transfer may have future clinical implications for Menkes disease patients with severe ATP7A mutations, as well as for patients with ATP7A-related motor neuron disease (see below).
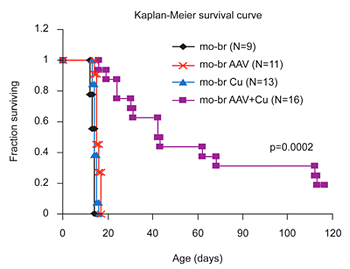
Figure 2. Brain-directed AAV5 gene therapy, in combination with copper, rescues a murine model of severe Menkes disease.
Markedly enhanced survival of a mouse model of severe Menkes disease: combined intracerebroventricular treatment using 1) AAV5 harboring a small version of the human ATP7A cDNA and 2) copper chloride. Gene therapy was administered on day 2 and copper on day 3. Neither treatment alone proved effective.
Distal hereditary motor neuropathies
Distal hereditary motor neuropathies (HMN) constitute a clinically and genetically heterogeneous group of disorders. Affected individuals manifest progressive weakness and wasting beginning in the distal muscles of the limbs, without notable sensory symptoms. Distal HMNs have been classified into seven subgroups based on mode of inheritance, age of onset, distribution of muscle weakness, and clinical progression. Fifteen genetic loci for distal HMN have been mapped, with eight genes identified to date. These encode a functionally diverse array of gene products including a transfer RNA synthetase, two heat shock proteins, and a microtubule motor protein involved in axonal transport. We recently identified an X-linked form of this disorder in three unrelated families associated with novel missense mutations in ATP7A. The three alterations affect highly conserved amino acids in the carboxyl half of ATP7A but do not directly involve the copper transporter’s critical functional domains. Functional characterization of mutant alleles included Western blotting, immunocytochemical analysis of cultured fibroblasts, and yeast complementation assays. Our results indicated normal ATP7A mRNA and protein levels, a temperature-sensitive defect in ATP7A trafficking, and partial rescue of a S. cerevisiae copper transport knockout. While ATP7A defects are typically associated with severe Menkes disease, or its milder allelic variant, occipital horn syndrome, we demonstrated that certain missense mutations at this locus can cause a syndrome restricted to progressive distal motor neuropathy without overt signs of systemic copper deficiency. This novel genotype-phenotype correlation suggests an important role of the ATP7A copper transporter in motor neuron maintenance and function. Our immediate plans include creation and evaluation of a knock-in mouse model, additional analyses of ATP7A function and trafficking in cultured cells from affected patients, expression of the mutant alleles in a mouse motor neuro–like cell line (NSC-34), and clinical evaluation of additional affected patients (under protocol #09-CH-0059).
Occipital horn syndrome
The synthetic amino acid L-threo-3,4-dihydroxyphenylserine (L-DOPS) is highly effective in reversing neurogenic orthostatic hypotension and correcting neurochemical abnormalities in patients with autosomal recessive congenital absence of dopamine-beta-hydroxylase (DBH). L-DOPS is metabolized by the enzyme DOPA decarboxylase to produce norepinephrine, thus bypassing the DBH enzymatic defect. Individuals with occipital horn syndrome (OHS) have partial deficiency of DBH, which is copper-dependent, and show distinctive neurochemical abnormalities and often symptoms of dysautonomia, such as syncope, dizziness, orthostatic hypotension, abnormal sinoatrial conduction, nocturnal bradycardia, and bowel or bladder dysfunction. As in patients with congenital absence of DBH, these problems in autonomic nervous system function seem potentially responsive to restoration of normal neurochemical levels. We hypothesize that L-DOPS treatment in OHS patients with dysautonomia will correct or improve blood neurochemical levels, and produce concurrent symptomatic improvement. A recently IRB-approved pilot study will test this hypothesis in six patients with dysautonomic symptoms, by providing L-DOPS treatment (5mg/kg/d po qd) and assessing the neurochemical response (in-hospital plasma catechol levels) and the longer-term effect on symptoms (out-of-hospital autonomic symptom questionnaire completed weekly by subject and/or parent), during an eight-week period while receiving the study drug.
Patent filed
Patent 4239-81164-01, Identification of subjects likely to benefit from copper treatment. International Filing Date: 06 October 2008
Clinical protocols
- 90-CH-0149: Early Copper Histidine Treatment in Menkes Disease.
- 09-CH-0059: Molecular Bases of Response to Copper Treatment in Menkes Disease, Related Phenotypes, and Unexplained Copper Deficiency.
- Protocol # pending: Pilot Study of L-DOPS for Dysautonomia in Occipital Horn Syndrome.
Publications
- Kaler SG, Holmes CS, Goldstein DS, Tang JR, Godwin SC, Donsante A, Liew CJ, Sato S, Patronas N. Neonatal diagnosis and treatment of Menkes disease. N Engl J Med 2008 358:605-614.
- Tang J, Donsante A, Desai V, Patronas N, Kaler SG. Clinical outcomes in Menkes disease patients with a copper-responsive ATP7A mutation, G727R. Molec Genet Metab 2008 95:174-182.
- Desai V, Kaler SG. Role of copper in human neurological disorders. Am J Clin Nutr 2008 88:855S-888S.
- Goldstein DS, Holmes CS, Kaler SG. Relative efficiencies of plasma catechol levels and ratios for neonatal diagnosis of Menkes disease. Neurochem Res 2009 34:1464-1468.
- Kaler SG, Tang J, Donsante A, Kaneski CR. Translational read-through of a nonsense mutation in ATP7A impacts treatment outcome in Menkes disease. Ann Neurol 2009 65:108-113.
Collaborators
- James Y. Garbern, MD PhD, Wayne State University School of Medicine
- David S. Goldstein, MD, PhD, Clinical Neurosciences Program, NINDS, Bethesda, MD
- Courtney Holmes, CMT, Clinical Neurosciences Program, NINDS, Bethesda, MD
- Marina L. Kennerson, PhD, University of Sydney, Sydney, Australia
- Clarissa Jang Liew, MD, Epilepsy Research Branch, NINDS, Bethesda, MD
- Julian F. Mercer, PhD, Deakin University, Melbourne, Australia
- Garth A. Nicholson, MD, PhD, University of Sydney, Sydney, Australia
- Nicholas Patronas, MD, Diagnostic Radiology Department, Clinical Center, NIH, Bethesda, MD
- Susumu Sato, MD, Epilepsy Research Branch, NINDS, Bethesda, MD
- Peter Steinbach, PhD, Center for Molecular Modeling, CIT, NIH, Bethesda, MD
- Vincent Timmerman, PhD, Universiteit Antwerpen, Antwerpen, Belgium