


You are here: Home > Unit on Pediatric Genetics
Translational Research on Mammalian Copper Transport Disorders

- Stephen G. Kaler, MD, MPH, Head, Unit on Human Copper Metabolism
- Anthony Donsante, PhD, Postdoctoral Fellow
- Ling Yi, PhD, Postdoctoral Fellow
- Julia D. Hicks, BS, Postbaccalaureate Fellow
The Unit on Human Copper Metabolism (formerly the Unit on Pediatric Genetics) conducts translational research focusing on the diagnosis, treatment, and understanding of copper metabolism disorders, a recently expanded collection of human diseases that impact the central and peripheral nervous systems. Menkes disease, the prototype genetic syndrome of defective copper transport, is characterized by low brain copper levels, infantile neurodegeneration, and premature death. The condition is caused by mutations in ATP7A, an X-linked gene that encodes a P-type copper ATPase. The occipital horn syndrome (OHS) is a milder allelic variant of Menkes disease, in which autonomic dysfunction and connective tissue abnormalities predominate. OHS is often associated with leaky splice junction mutations. We and our colleagues recently identified two families with a distal motor neuropathy resembling Charcot-Marie-Tooth disease type 2, which is caused by novel missense mutations (Thr994Ile and Pro1386Ser) in or near transmembrane segments of ATP7A. The mutations are associated with abnormal intracellular trafficking of ATP7A. Thus, the phenotypic spectrum of ATP7A mutations has expanded; discovery of this new allelic variant also indicates a previously unappreciated role for ATP7A in motor neuron maintenance and function, which the Unit hopes to unravel.
We are also working on the delineation of a possible new autosomal recessive syndrome caused by mutations in a copper chaperone gene and which implies a link between cellular apoptosis and copper transport.
Gene replacement in a mouse model of Menkes disease
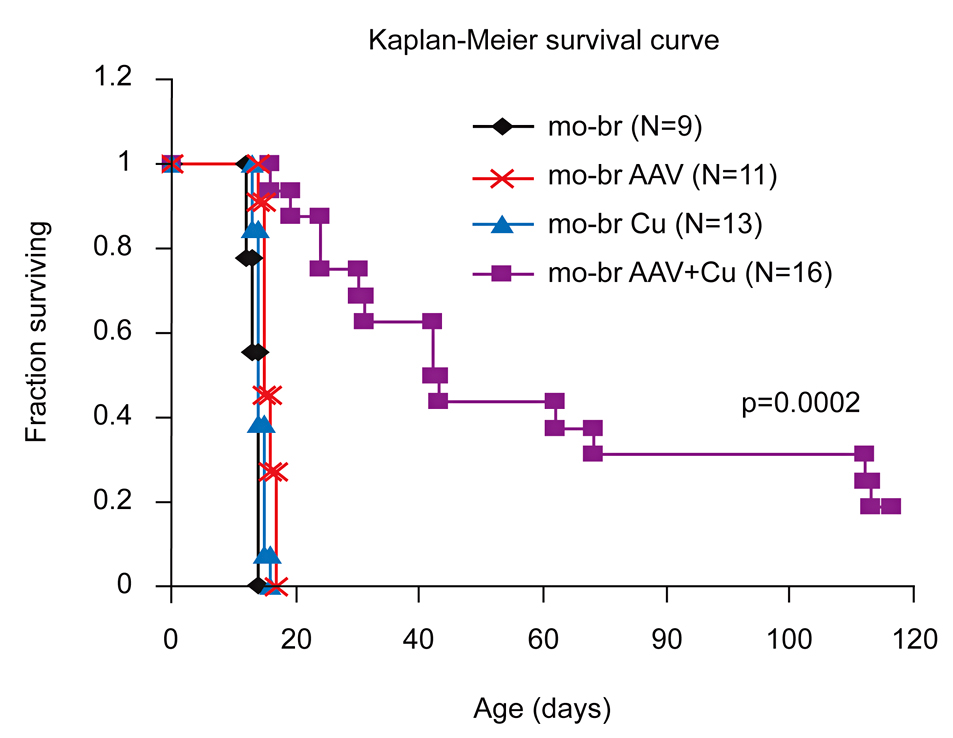
Figure 1. Brain-directed AAV5 gene therapy, in combination with copper, rescues a murine model of severe Menkes disease.
Markedly enhanced survival of a mouse model of severe Menkes disease: combined intracerebroventricular treatment using 1) AAV5 harboring a small version of the human ATP7A cDNA and 2) copper chloride. Gene therapy was administered on day 2 and copper on day 3. Neither treatment alone proved effective. (click image to enlarge)
The mottled-brindled (mo-br) mouse manifests a lethal abnormality in copper transport to the brain caused by a mutation in atp7a, a P-type ATPase, and is a model for Menkes disease. We treated neonatal mo-br mice with either intracerebroventricular recombinant adeno-associated virus serotype 5 (AAV) harboring an atp7a homolog, intracerebroventricular copper, or both. Only AAV+Cu treatment rescued mo-br (Figure 1). At the viral titers (5x108 vector genomes) employed, transduction by AAV occurred primarily in the choroid plexus epithelia. Survival was associated with higher brain copper levels and improved activity of dopamine-beta-hydroxylase, a copper enzyme metallated in the trans-Golgi compartment. In contrast, the activities of cytosolic and mitochondrial copper enzymes did not differ notably among treatment groups.
We performed weekly serial neurobehavioral testing in long-surviving mo-br mice using the constant speed rotarod (balance and coordination) and wire hang (neuromuscular strength) beginning at 25d of age up to 300d of age (3 trials per time point, 60s maximum). AAV+Cu mo-br mice performed similarly to wild-type controls on the rotarod at 25d of age (mo-br [N=10] 56s, wild type [N=9] 60s). However, they exhibited significant impairment in later weeks, averaging 15-20s on the rod. Surprisingly, three AAV+Cu mo-br mice regained the ability to remain on the rotarod for the full 60s beginning at ages 95d, 144d, and 173d. On the wire hang test, AAV+Cu mice performed abnormally at 25d of age (30s vs. wild type 60s; p<10-4) but showed improvement with age. At 116d, mo-br mice (N=6) stayed on the wire for a mean duration of 54s versus 60s in wild type (N=10). Of AAV+Cu mo-br mice 67% completed the full 60s at this time point compared with 10% at 25d of age. Electron micrographs showed no overt ultrastructural abnormalities and myelin staining appeared similar in 300-day-old treated mo-br brain and normal controls.
Our findings suggest that 1) rescue of the mo-br mouse is dependent upon improved copper retention in the brain due to transduction of choroid plexus epithelia, 2) copper enzymes metallated in the trans-Golgi network are responsible for improved life span in AAV+Cu mo-br mice, and 3) recovery of motor function in the rotarod and wire hang test long after administration of therapy may be explained by slower than normal rates of myelination and axonal growth/development.
Mechanism of neurodegeneration in ATP7A-related distal motor neuropathy
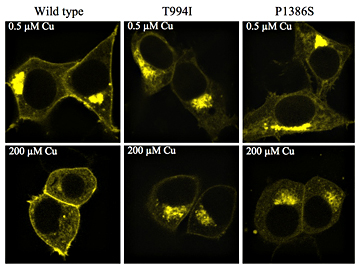
Figure 2. Mutant ATP7A alleles causing distal motor neuropathy traffic abnormally.
Confocal images of HEK-293 cells transfected with plasmids harboring ATP7A alleles tagged with Venus-fluorescent protein. Compared with the wild-type allele, the ATP7A trafficking response after exposure to increased copper (200 µM for 3 h) is delayed for the Thr994Ile and Pro1386Ser mutant alleles.
Distal hereditary motor neuropathies (HMN) constitute a clinically and genetically heterogeneous group of disorders. Whereas Menkes disease and occipital horn syndrome (OHS), the milder allelic variant, share certain clinical and biochemical abnormalities, recently identified subjects with ATP7A-related distal motor neuropathy have normal serum copper, normal copper enzyme activities, normal renal tubular function, no central nervous system symptoms, and no connective tissue abnormalities. Conversely, three Menkes disease patients and four OHS patients whom we studied recently showed no clinical or neurophysiological evidence of motor neuron dysfunction, including the original OHS patient we reported on, who is now 32, an age by which distal motor neuropathy would be expected to manifest itself. Together, these clinical findings imply that the mechanism of disease in the new allelic variant is distinct from that in Menkes/OHS.
To characterize the unique missense mutations that cause ATP7A-related distal motor neuropathy, we expressed Pro1386Ser and Thr994Ile in a yeast copper transport knockout, ccc2delta, as well as in Hek293 cells, in a fibroblast cell line from a Menkes patient with a large ATP7A deletion, and in motor neuron–enriched NSC-34 cells. For mammalian cell transfections, we tagged wild-type and mutant ATP7A alleles with Venus-fluorescent protein. The mutant alleles complemented ccc2delta in the range of 60-70% of normal. Expression in Hek293 cells suggested delayed trafficking of mutant ATP7As in response to copper loading (Figure 2), as noted previously in fibroblasts from affected patients. In addition, we observed accumulation of mutant ATP7As at the plasma membrane. This phenomenon was clearer in transfected neuronal (NSC-34) cells in which mutant ATP7A also showed more diffuse intracellular localization than in wild type. The results suggest that aberrant ATP7A trafficking, particularly relevant when transport along lengthy axons is required, may sabotage motor neuron function and underlie this phenotype. Protein misfolding with oligomerization or aggregate formation, chronic deficiency of one or more copper enzyme(s), oxidative injury, and altered synaptic activity of ATP7A remain alternative potential disease mechanisms. Delineation of the problem in this form of distal motor neuropathy will help elucidate the normal role of ATP7A in motor neurons.
Somatic mosaicism in Menkes disease suggests choroid plexus-mediated copper transport to the developing brain.
The primary mechanism of copper transport to the brain is unknown, although this process is drastically impaired in Menkes disease. Potential central nervous system entry routes for copper include brain capillary endothelial cells, which originate from mesodermal angioblasts and form the blood-brain barrier, and the choroid plexuses, which derive from embryonic ectoderm and form the blood-cerebrospinal fluid barrier. We exploited a rare (and first reported) example of somatic mosaicism for an ATP7A mutation to shed light on questions about copper transport into the developing brain. In a 20-month-old Menkes disease patient evaluated before copper treatment, blood copper and catecholamine concentrations were normal, whereas levels in cerebrospinal fluid were abnormal and consistent with his neurologically severe phenotype. We documented disparate levels of mosaicism for an ATP7A missense mutation, P1001L, in tissues derived from different embryonic origins; allele quantitation showed P1001L in approximately 27% and 88% of DNA samples from blood cells (mesoderm-derived) and cultured fibroblasts (ectoderm-derived), respectively. The findings imply that the P1001L mutation in the patient preceded formation of the three primary embryonic lineages at gastrulation, with the ectoderm layer ultimately harboring a higher percentage of mutation-bearing cells than mesoderm or endoderm. Given that choroid plexus epithelia are derived from neuroectoderm and brain capillary endothelial cells from mesodermal angioblasts, the clinical, biochemical, and molecular findings in this child support a critical role for the blood-CSF barrier (choroid plexus epithelia) in copper entry to the developing brain.
Droxidopa for dysautonomia in occipital horn syndrome and Menkes disease
Individuals with occipital horn syndrome (OHS), as well as Menkes disease patients who survive due to neonatal diagnosis and early copper treatment, often have a partial deficiency of dopamine-beta-hydroxylase (DBH) and show distinctive neurochemical abnormalities and symptoms of dysautonomia, such as syncope, dizziness, orthostatic hypotension, abnormal sinoatrial conduction, nocturnal bradycardia, and bowel or bladder dysfunction. As in patients with congenital absence of DBH, the problems in autonomic nervous system function seem potentially responsive to restoration of normal neurochemical levels.
We arranged a clinical trial agreement with Chelsea Therapeutics, Raleigh, NC, to obtain the synthetic amino acid l-threo-3,4-dihydroxyphenylserine (Droxidopa). Droxidopa is highly effective in reversing neurogenic orthostatic hypotension and correcting neurochemical abnormalities in patients with autosomal recessive congenital absence of DBH. Droxidopa is metabolized by the enzyme DOPA decarboxylase to produce norepinephrine, thus bypassing the DBH enzymatic defect. We hypothesized that Droxidopa treatment in OHS and Menkes patients with dysautonomia would correct or improve blood neurochemical levels, and produce concurrent symptomatic improvement. A recently IRB-approved pilot study will test this hypothesis in six patients with dysautonomic symptoms by providing Droxidopa treatment (5mg/kg/d po qd) and assessing the neurochemical response (in-hospital plasma catechol levels) and the longer-term effect on symptoms (out-of-hospital autonomic symptom questionnaire completed weekly by subject and/or parent), during an eight-week period while receiving the study drug.
Molecular correlates of epilepsy in early-diagnosed and -treated Menkes disease
Epilepsy is a major feature of Menkes disease. Three prior surveys indicated clinical seizures and electroencephalographic (EEG) abnormalities in a combined 27 of 29 (93%) symptomatic Menkes disease patients diagnosed at 2 months of age or older. To assess the influence of early, presymptomatic diagnosis and treatment on seizure semiology and brain electrical activity, we evaluated 71 EEGs in 24 Menkes disease patients who were diagnosed and treated with copper injections in early infancy (≤6 weeks of age) and whose ATP7A mutations we determined. Clinical seizures were observed in only 12.5% (3/24) of these patients, although 46% (11/24) had at least one abnormal EEG tracing, including 50% of patients with large deletions in ATP7A, 50% of those with small deletions, 60% of those with nonsense mutations, and 57% of those with canonical splice junction mutations. In contrast, five patients with mutations shown to retain partial function, either via some correct RNA splicing or residual copper transport capacity, had neither clinical seizures nor EEG abnormalities. Our findings suggest that early diagnosis and treatment improve brain electrical activity and decrease seizure occurrence in classical Menkes disease, irrespective of the precise molecular defect. Subjects with ATP7A mutations that retain some function seem particularly well protected by early intervention against the possibility of epilepsy.
Clinical protocols
- Principal Investigator, 90-CH-0149 Early copper histidine treatment in Menkes disease: relationship of molecular defects to neurodevelopmental outcomes
- Associate Investigator, 02-CH-0023 Studies of pediatric patients with metabolic or other genetic disorders
- Principal Investigator, 09-CH-0059 Molecular bases of response to copper treatment in Menkes disease, related phenotypes, and unexplained copper deficiency
- Principal Investigator, 10-CH-xxxx Pilot study of Droxidopa for dysautonomia in Menkes disease and occipital horn syndrome
Patent filed
- Patent 4239-81164-01, Identification of subjects likely to benefit from copper treatment. International Filing Date: 06 October 2008
Publications
- Goldstein DS, Holmes CS, Kaler SG. Relative efficiencies of plasma catechol levels and ratios for neonatal diagnosis of Menkes disease. Neurochem Res. 2009; 34:1464-1468.
- Kaler SG, Tang J, Donsante A, Kaneski CR. Translational read-through of a nonsense mutation in ATP7A impacts treatment outcome in Menkes disease. Ann Neurol. 2009; 65:108-113.
- Kaler SG. ATP7A-related copper transport diseases—emerging concepts and future trends. Nat Rev Neurol. 2010; in press.
- Kennerson ML, Nicholson GA, Kaler SG, Kowalski B, Mercer JF, Tang J, Llanos RM, Chu S, Takata RI, Speck-Martins CE, Baets J, Almeida-Souza L, Fischer D, Timmerman V, Taylor PE, Scherer SS, Ferguson TA, Bird TD, De Jonghe P, Feely SM, Shy ME, Garbern JY. Missense mutations in the copper transporter gene ATP7A cause X-linked distal hereditary motor neuropathy. Am J Hum Genet. 2010; 86:343-352.
- Desai V, Donsante A, Swoboda KJ, Martensen M, Thompson J, Kaler SG. Favorably skewed X-inactivation accounts for neurological sparing in female carriers of Menkes disease. Clin Genet. 2010;Apr 19. [Epub ahead of print].
- Kaler SG, Liew CJ, Donsante A, Hicks JD, Sato S, Greenfield JC. Molecular correlates of epilepsy in early diagnosed and treated Menkes disease. J Inher Metab Dis. 2010; 33:583-589.
- Donsante A, Johnson P, Jansen LA, Kaler SG. Somatic mosaicism in Menkes disease suggests choroid plexus-mediated copper transport to the developing brain. Am J Med Genet A. 2010;152A:2529-2534.
- Kaler SG. ATP7A-Related Copper Transport Disorders. In: Pagon RA, Bird TC, Dolan CR, Stephens K, editors. GeneReviews [Internet]. Seattle (WA): University of Washington, Seattle, 1997-2003. http://www.genetests.org [updated 2010].
- Kaler SG. Small copper complexes for treatment of acquired and inherited copper deficiency syndromes. In Thoene J (ed), Small Molecule Therapy for Genetic Disease 2010; Cambridge University Press.
- Goldman L, Ausiello D, Arend W, Armitage JO, Clemmons D, Drazen J, Griggs R, LaRusso N, Newman J, Foster E (eds). Wilson disease. Cecil's Textbook of Medicine 24th edition 2010; Chapt. 218:in press.
- Dulac O, Sarnat H, Lassonde M, eds. Inborn errors of copper metabolism. Pediatric Neurology Handbook of Clinical Neurology, Third Series 2010; in press.
Collaborators
- James Y. Garbern, MD, PhD, Wayne State University School of Medicine
- David S. Goldstein, MD, PhD, Clinical Neurosciences Program, NINDS, Bethesda, MD
- Courtney Holmes, CMT, Clinical Neurosciences Program, NINDS, Bethesda, MD
- Peter Huppke, MD, Georg August Universität, Göttingen, Germany
- Marina L. Kennerson, PhD, University of Sydney, Sydney, Australia
- Clarissa Jang Liew, MD, Epilepsy Research Branch, NINDS, Bethesda, MD
- Julian F. Mercer, PhD, Deakin University, Melbourne, Australia
- Garth A. Nicholson, MD, PhD, University of Sydney, Sydney, Australia
- Nicholas Patronas, MD, Diagnostic Radiology Department, Clinical Center, NIH, Bethesda, MD
- Susumu Sato, MD, Epilepsy Research Branch, NINDS, Bethesda, MD
- Peter Steinbach, PhD, Center for Molecular Modeling, CIT, NIH, Bethesda, MD
- Vincent Timmerman, PhD, Universiteit Antwerpen, Antwerpen, Belgium