

You are here: Home > Unit on Pediatric Genetics
Gene Therapy for Neurometabolic Disorder

- Stephen G. Kaler, MD, MPH, Head, Unit on Human Copper Metabolism
- Anthony Donsante, PhD, Postdoctoral Fellow
- Ling Yi, PhD, Postdoctoral Fellow
- Julia D. Hicks, BS, Postbaccalaureate Fellow
- Paul Johnson, BS, Medical Student
We are committed to identifying the genetic causes of copper transport diseases, understanding how the responsible genes participate in neurologic processes, dissecting disease mechanisms, and using this knowledge to improve health through rational treatments, including gene therapy. Patients and families affected by inherited neurometabolic disorders provide the impetus for scientific inquiry. In addition to molecular genetics, the laboratory employs model organisms (yeast, mouse), cellular, biochemical, and biophysical approaches, and human clinical trials.

Kaler Lab photo
(L-R) Johnson, Hicks, Donsante, Yi
ATP7A gene therapy in murine models of Menkes disease
Click image to enlarge.
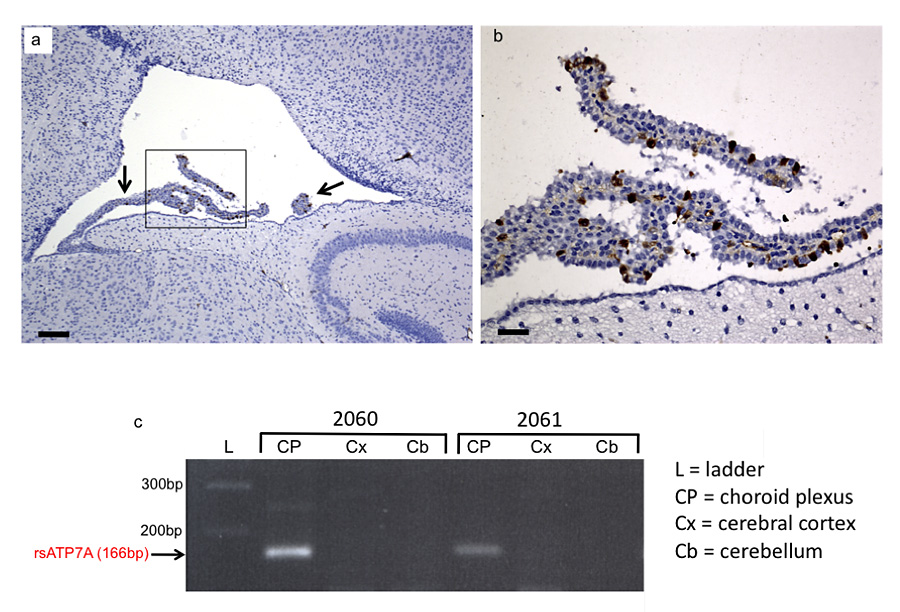
Figure 1. Selective transduction of choroid plexus epithelia
(a) Transduction of choroid plexus epithelia by AAV5-GFP. Low power (scale bar = 160 μm) and (b) high power (scale bar = 40 μm) views of the lateral ventricle and choroid plexus (arrow) from a 12-day-old wild-type mouse brain stained with anti-GFP (brown). The same transduction pattern was evident in AAV5-GFP+Cu–treated mo-br mice. (c) Reverse transcriptase (RT)-PCR confirms selective choroid plexus transduction by AAV5-rsATP7A. RT-PCR in mice 2060 and 2061 shows amplification of the 166 bp expected transgene product only in choroid plexus RNA samples.
Menkes disease is a lethal infantile neurodegenerative disorder of copper metabolism caused by mutations in a P-type ATPase, ATP7A. Currently available treatment is not entirely effective in a majority of affected individuals, and mortality is high. The mottled-brindled (mo-br) mouse recapitulates the Menkes phenotype, including abnormal copper transport to the brain owing to mutation in the murine homolog Atp7a, and dies by 14 days of age. We documented that mo-br mice on a C57BL/6 background were not rescued by peripheral copper administration, and we used this model to evaluate brain-directed therapies. Neonatal mo-br mice received lateral ventricle injections of either adeno-associated virus serotype 5 (AAV5) harboring a reduced-size (rs) human ATP7A (rsATP7A) complementary DNA (cDNA), copper chloride, or both. AAV5-rsATP7A showed selective transduction of choroid plexus epithelia (Figure 1) and AAV5-rsATP7A plus copper combination treatment rescued mo-br mice; 86% survived to weaning (21 days), median survival increased to 43 days, 37% lived beyond 100 days, and 22% survived to the study end point (300 days). This synergistic treatment effect correlated with increased brain copper levels, enhanced activity of dopamine-beta-hydroxylase, a copper-dependent enzyme, and correction of brain pathology. These findings provide the first definitive evidence that gene therapy may have clinical utility in the treatment of Menkes disease.
AAV9 gene therapy
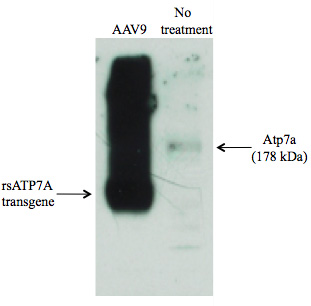
Figure 2. Robust AAV9–mediated expression of rsATP7A in mouse skeletal muscle compared with endogenous Atp7a (178 kDa) in an untreated animal
Systemic adeno-associated virus (AAV) gene therapy could enable safer, less invasive delivery than brain-directed approaches. To refine prospects for clinical translation of gene therapy for Menkes disease, we evaluated transduction efficiency and therapeutic effectiveness of AAV serotype 9, which has a unique capacity to cross the blood-brain and blood-CSF barriers. In a cohort of mo-br mice, we administered 5x1011 viral genome particles of AAV9 harboring the rs human ATP7A cDNA by intraperitoneal injection on day 2 of life. Western blot analysis indicated high rsATP7A transgene expression in skeletal muscle (Figure 2), and transduction of liver, kidney, heart, lung, intestine, cerebral cortex and choroid plexus epithelia was confirmed by quantitative PCR. Median life span was twice that of untreated affected mo-br mice, demonstrating the efficacy of systemic AAV9. The AAV9 serotype may offer even more effective long-term outcomes than AAV5, based on broader neuronal tropism. We also performed successful in utero AAV9 gene transfer in E15 mouse embryos, with a view to treatment of the dappled mouse allele, a large intragenic deletion in Atp7a that results in prenatal lethality.
Capsid engineering for choroid plexus–specific AAV transduction
The choroid plexus (CP) is a highly vascularized structure that projects into the ventricles of the brain. Besides creating the blood-CSF barrier, the polarized epithelia of the choroid plexus produce CSF by transporting water and ions into the ventricles from the blood and secreting a large number of proteins. Several neuro-metabolic diseases, such as lysosomal storage disorders, could benefit from a CP–targeted gene therapy approach, given that CSF flow carries molecules throughout the ventricular system into the subarachnoid space, which covers the entire brain surface. Based on our observation that selective transduction of CP epithelia enabled rescue of the mo-br mouse, we are developing an AAV vector selectively evolved to target these specialized cells, via phage panning. This powerful method has been used to identify viral capsid peptide motifs that allow superior vector homing to specific cells and tissues. Ultimately, we will evaluate the capacity of CP–specific, selectively evolved AAV to refine and improve outcomes in the mo-br mouse.
Choroid plexus–directed gene therapy for Sanfilippo B syndrome
In collaboration with Patricia Dickson, we embarked on a study of CP–directed gene therapy for mucopolysaccharidosis type IIIB (Sanfilippo B syndrome), a devastating neurological disorder caused by N-acetylglucosaminidase (NAGLU) deficiency. Sanfilippo B is a prototype for a broader category of neurometabolic disorders for which brain-directed gene therapy may prove paradigm-shifting in the treatment of lysosomal storage diseases. No specific therapy is currently available for Sanfilippo B, and disease management consists solely of supportive care. The development of enzyme replacement therapy has been hampered by poor mannose 6-phosphorylation of NAGLU when produced in recombinant form. However, Dickson's laboratory developed a fusion protein consisting of insulin-like growth factor 2 (IGF2) and NAGLU that independently binds to the mannose 6-phosphate receptor and shows better intracellular uptake by Sanfilippo B fibroblasts than does wild-type recombinant NAGLU. This is because the IGF2 moiety binds to the mannose 6-phosphate receptor independently of mannose 6-phosphorylation.
Intrathecal delivery of the recombinant enzyme (injecting enzyme into the cerebrospinal fluid during a spinal tap) has been successful in animal models of other lysosomal storage diseases. However, a major drawback to this approach is the need for repeated (e.g., monthly) intrathecal injections. An alternative route of administration would be transduction of CP epithelial cells with an AAV vector containing the cDNA for the enzyme of interest. Given that CP epithelia have an extremely low rate of turnover, this approach would create a "permanent source" of enzyme in the CSF for utilization by the brain. Our collaborative studies, recently co-funded by the National MPS Society, will address this issue for Sanfilippo B syndrome.
Novel disease mechanisms underlie ATP7A–related distal motor neuropathy
The P-type ATPase ATP7A regulates cellular copper homeostasis by its activity at the trans-Golgi network (TGN) and plasma membrane (PM), with its location normally governed by intracellular copper concentration. In addition to causing Menkes disease, defects in ATP7A may lead to the disease variants occipital horn syndrome and ATP7A–related distal motor neuropathy, a newly discovered condition for which the precise pathophysiology has been obscure. We characterized two ATP7A motor neuropathy mutations (T994I, P1386S) previously associated with abnormal intracellular trafficking. In the patients' fibroblasts, total internal reflection fluorescence (TIRF) microscopy indicated a shift in steady-state equilibrium of ATP7AT994I and ATP7AP1386S, with excess PM localization. Transfection of 293T cells and NSC-34 motor neurons with the mutant alleles tagged with Venus fluorescent protein also showed higher PM localization. Endocytic retrieval of the mutant alleles from the PM to the TGN was delayed. Immunoprecipitation assays revealed an abnormal interaction between ATP7AT994I and p97/VCP, a TGN–resident protein associated with two other inherited motor neuropathies, including amyotrophic lateral sclerosis. SiRNA knockdown of p97/VCP improved ATP7AT994I localization. Our findings illuminate mechanisms underlying ATP7A–related distal motor neuropathy, establish a common link between genetically distinct forms of motor neuron disease, clarify the normal process of ATP7A endocytosis, and highlight possible functional roles of ATP7A in the peripheral nervous system.
Clinical Protocols
- Principal Investigator, 90-CH-0149 Early copper histidine treatment in Menkes disease: relationship of molecular defects to neurodevelopmental outcomes
- Associate Investigator, 02-CH-0023 Studies of pediatric patients with metabolic or other genetic disorders
- Principal Investigator, 09-CH-0059 Molecular bases of response to copper treatment in Menkes disease, related phenotypes, and unexplained copper deficiency
Patent filed
Patent 4239-81164-01, Identification of subjects likely to benefit from copper treatment. International Filing Date: 06 October 2008
Additional Funding
- NICHD, National MPS Society
Publications
- Donsante A, Yi L, Zerfas P, Brinster L, Sullivan P, Goldstein DS, Prohaska J, Centeno JA, Kaler SG. ATP7A gene addition to the choroid plexus results in long-term rescue of the lethal copper transport defect in a Menkes disease mouse model. Mol Ther 2011;doi:10.1038/mt.2011.143.
- Kaler SG. ATP7A-related copper transport diseases—emerging concepts and future trends. Nat Rev Neurol 2011;7:15-29.
- Kennerson ML, Nicholson GA, Kaler SG, Kowalski B, Mercer JF, Tang J, Llanos RM, Chu S, Takata RI, Speck-Martins CE, Baets J, Almeida-Souza L, Fischer D, Timmerman V, Taylor PE, Scherer SS, Ferguson TA, Bird TD, De Jonghe P, Feely SM, Shy ME, Garbern JY. Missense mutations in the copper transporter gene ATP7A cause X-linked distal hereditary motor neuropathy. Am J Hum Genet 2010;86:343-352.
- Desai V, Donsante A, Swoboda KJ, Martensen M, Thompson J, Kaler SG. Favorably skewed X-inactivation accounts for neurological sparing in female carriers of Menkes disease. Clin Genet 2011;79:176-182.
- Donsante A, Johnson P, Jansen LA, Kaler SG. Somatic mosaicism in Menkes disease suggests choroid plexus-mediated copper transport to the developing brain. Am J Med Genet A 2010;152A:2529-2534.
Collaborators
- Patricia Dickson, MD, Harbor-UCLA, Los Angeles, California
- James Y. Garbern, MD, PhD, Wayne State University School of Medicine, Detroit, MI
- David S. Goldstein, MD, PhD, Clinical Neurosciences Program, NINDS, Bethesda, MD
- Courtney Holmes, CMT, Clinical Neurosciences Program, NINDS, Bethesda, MD
- Peter Huppke, MD, Georg August Universität, Göttingen, Germany
- Marina L. Kennerson, PhD, University of Sydney, Sydney, Australia
- Nicholas Patronas, MD, Diagnostic Radiology Department, NIH Clinical Center, Bethesda, MD
- Peter Steinbach, PhD, Center for Molecular Modeling, CIT, NIH, Bethesda, MD