

You are here: Home > Section on Medical Neuroendocrinology
Diagnosis, Localization, Pathophysiology, and Molecular Biology of Pheochromocytoma and Paraganglioma

- Karel Pacak, MD, PhD, DSc, Head, Section on Medical Neuroendocrinology
- Alessio Giubellino, MD, Senior Research Fellow
- Thanh-Truc Huynh, MS, Biologist
- Karen T. Adams, MSc, CRNP, Research Nurse
- Jan Schovanek, MD, Postdoctoral Visiting Fellow
- Jaydira Del Rivero, MD, Clinical Fellow
- Nikoletta Lendvai, MD, Volunteer
- Ivana Jochmanova, MS, Volunteer
- Petra Bullova, MS, Predoctoral Visiting Fellow
- Victoria Martucci, BA, Postbaccalaureate Fellow
We conduct patient-oriented research into the etiology, pathophysiology, genetics, diagnosis, localization, and treatment of pheochromocytoma and paraganglioma. Projects include both translational research—applying basic science knowledge to clinical diagnosis, pathophysiology, and treatment—and “reverse translation research,” by which clinical findings lead to new concepts for pursuit by basic researchers in the laboratory. Our goals are to: (i) establish new and improved methods and strategies for novel diagnostic and localization approaches to pheochromocytoma/paraganglioma; (ii) explain the molecular and cellular basis for varying clinical presentations of pheochromocytomas/paragangliomas and establish the pathways of tumorigenesis; (iii) search for new molecular and genetic markers for diagnosis and treatment of metastatic pheochromocytoma/paraganglioma; (iv) introduce new therapeutic options for malignant/metastatic pheochromocytoma/paraganglioma; and (v) facilitate new and improved collaborations and interdisciplinary studies. To achieve these goals, we base our strategy on multidisciplinary collaborations with investigators from several NIH Institutes and outside medical centers. We link a patient-oriented component with two bench-level components. The patient-oriented component (medical neuroendocrinology) is the driving force for our hypotheses and discoveries. The two bench-level components (tumor pathogenesis/genetics and chemistry; biomarkers) emphasize, first, technologies of basic research tailored for pathway and target discovery and, second, the further development of discoveries into clinical applications.
Hereditary pheochromocytoma and paraganglioma
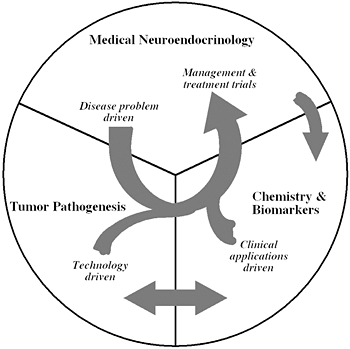
Figure 1.
The unique structure of our Section tightly links a patient-oriented component with two bench-level components. One of the latter two components emphasizes new technologies of basic research tailored for pathway and target discovery; the other focuses on further development of those and other discoveries into clinical applications.
Many solid tumors, including pheochromocytoma and paraganglioma, are characterized by a (pseudo)hypoxic signature. (Pseudo)hypoxia has been shown to promote both tumor progression and resistance to therapy. The major mediators of the transcriptional hypoxic response are hypoxia-inducible factors (HIFs) (1). High levels of HIFs lead to transcription of hypoxia-responsive genes, which are involved in tumorigenesis. Pheochromocytomas and paragangliomas are catecholamine-producing tumors arising from sympathetic- or parasympathetic-derived chromaffin tissue. In recent years, substantial progress has been made in understanding the metabolic disturbances present in pheochromocytoma and paraganglioma, especially because of the identification of some disease-susceptibility genes. To date, fifteen pheochromocytoma and paraganglioma susceptibility genes have been identified. Based on the main transcription signatures of the mutated genes, pheochromocytomas and paragangliomas have been divided into two clusters, pseudohypoxic cluster 1 and pseudohypoxic cluster 2, rich in kinase receptor signaling and protein translation pathways. Although the two clusters appear to show distinct signaling pathways, recent data suggest that both clusters are interconnected by HIF signaling as the important driver in their tumorigenesis, and mutations in most pheochromocytoma- and paraganglioma-susceptibility genes seem to affect HIFα regulation and its downstream signaling pathways. HIF signaling appears to play an important role in the development and growth of pheochromocytomas and paraganglioma, which could suggest new therapeutic approaches for the treatment of these tumors
No HIF2A mutations have been previously identified in any cancer. First, we reported two novel somatic gain-of-function HIF2A mutations in two patients, one presenting with multiple paragangliomas and a second with both multiple paragangliomas and multiple duodenal somatostatinoma, both associated with polycythemia (2). Both mutations showed increased HIF2alpha activity and protein half-life. While germline mutations of HIF2alpha regulators, including VHL, EGLN1, SDHB, SDHC, and SDHD, have been reported in pheochromocytomas/paragangliomas, this was the first report of a somatic gain-of-function mutation in HIF2A (2). Subsequently, we investigated two additional unrelated patients and found them to present with the same disease cluster (3). Our findings indicate the existence of a new syndrome with multiple paragangliomas and somatostatinomas associated with polycythemia. The new syndrome results from somatic gain-of-function HIF2A mutations, which cause an upregulation of hypoxia-related genes, including erythropoietin and genes important in cancer biology.
Pheochromocytoma/paraganglioma-related to mutations in the mitochondrial SDH subunits A, B, C, and D and in SDHAF-2 and VHL genes share a pseudohypoxic expression profile. However, genotype-specific differences in expression have been emerging. Development of effective new therapies for distinctive clinical features, e.g., the high rate of malignancy in SDHB patients or the predisposition to multifocal PGLs in SDHD patients mandates new stratification tools. To identify mutation/location-related characteristics among pseudohypoxic pheochromocytomas/paragangliomas, we used comprehensive microarray gene expression profiling (SDHB: n=18, SDHD-abdominal/thoracic [AT]: n=6, SDHD-head/neck [HN]: n=8, VHL: n=13). To avoid location-specific expression bias, we derived typical adrenal medulla genes from matched normal medullae and cortices (n=8) for data normalization. Unsupervised analysis identified two dominant clusters, separating SDHB and SDHD-AT PHEOs/PGLs (cluster A) from VHL pheochromocytomas and SDHD-HN paragangliomas (cluster B). Supervised analysis yielded 6,937 highly predictive genes (misclassification error rate 0.175). Enrichment analysis revealed that genes related to energy metabolism and inflammation/fibrosis were changed most in clusters A and B, respectively. A minimum subset of 40 classifiers was validated by quantitative RT-PCR (qRT-PCR vs. microarray: r = 0.87). Expression of several individual classifiers was identified as characteristic for VHL and SDHD-HN pheochromocytomas/paragangliomas, respectively. We showed for the first time that SDHD-HN paragangliomas share more features with VHL pheochromocytomas than with SDHD-AT paragangliomas. This suggests a novel sub-classification of pseudohypoxic pheochromocytomas/paragangliomas and implies cluster-specific pathogenic mechanisms and the need for treatment strategies targeted accordingly.
Imaging of various pheochromocytomas and paragangliomas
We performed a study aimed at establishing the sensitivity and specificity of 18F-fluorodihydroxyphenylalanine (18F-FDOPA) PET in relation to tumor localization and the patient’s genetic status in a large series of pheochromocytoma/paraganglioma patients and at assessing in detail false-negative results. A retrospective study of pheochromocytoma/paraganglioma patients investigated with 18F-FDOPA PET or PET/CT imaging in two academic endocrine tumor centers was conducted (La Timone University Hospital, Marseilles, France, and NIH, Bethesda, MD, USA). One hundred and sixteen patients (39.7% harboring germline mutations in known disease susceptibility genes) were evaluated for a total of 195 pheochromocytoma/paraganglioma foci. 18F-FDOPA PET correctly detected 179/195 lesions (pooled sensitivity was 91.8%) in 107 patients. All but one of the sporadic tumors were 18F-FDOPA–avid. 18F-FDOPA PET failed to detect 16/195 tumors in nine patients (5 SDHD, 3 SDHB, 1 sporadic). Lesion-based sensitivities for parasympathetic PGL (head, neck, or anterior/middle thoracic), pheochromocytoma, and extra-adrenal sympathetic (abdominal or posterior thoracic) paraganglioma were 98.2%, 93.9%, and 70.3%, respectively (P<0.001). All but two of the missed lesions were in the sympathetic region, and all patients with 18F-FDOPA–negative tumors had SDHx mutations. 18F-FDOPA PET failed to detect two head and neck paragangliomas, likely owing to their small size, while most missed sympathetic paraganglioma were larger and may have exhibited a specific 18F-FDOPA–negative imaging phenotype. 18F-fluorodeoxyglucose (18F-FDG) PET detected all the missed sympathetic lesions. We concluded that 18F-FDOPA PET appears to be a highly sensitive functional imaging tool for head and neck paragangliomas regardless of the genetic status of the tumors. Patients with false-negative sympathetic tumors on 18F-FDOPA PET should be tested for SDHx mutations.
Metastatic pheochromocytoma and paraganglioma
Metastatic pheochromocytoma/paraganglioma represents one of the major clinical challenges in the field of neuroendocrine oncology. Recent molecular characterization of pheochromocytoma/paraganglioma suggests new treatment options with targeted therapies. In one study, we investigated the 90 kDa heat shock protein (Hsp90) as a potential therapeutic target for advanced pheochromocytoma/paraganglioma. Both the first-generation natural Hsp90 inhibitor 17-allylamino-17-demethoxygeldanamycin (17-AAG, tanespimycin) and the second-generation synthetic Hsp90 inhibitor STA-9090 (ganetespib) demonstrated potent inhibition of proliferation and migration of pheochromocytoma cell lines and induced degradation of key Hsp90 clients. Furthermore, ganetespib induced dose-dependent cytotoxicity in primary pheochromocytoma cells. Using metastatic models of pheochromocytoma, we demonstrated the efficacy of 17-AAG and ganetespib in reducing metastatic burden and increasing survival. Levels of Hsp70 in plasma from the xenograft studies served as a proximal biomarker of drug treatment (5). Our study suggests that targeting Hsp90 may benefit patients with advanced pheochromocytoma/paraganglioma.
Another major deficiency in current chemotherapy strategies derived from the fact that that metastatic pheochromocytomas/paragangliomas usually grow very slowly, and most of the cells are quiescent at any given time. Drugs that target dividing tumor cells have, therefore, met with limited success. The need to improve treatment of metastatic pheochromocytoma/paraganglioma requires new strategies and a valid experimental model for pre-clinical testing. However, development of such a model has itself been hampered by the absence of human pheochromocytoma/paraganglioma cell lines for culture or xenograft studies. Topoisomerase 1 (TOP1) inhibitors are drugs that interfere with mechanisms that maintain DNA integrity during transcription in both quiescent and dividing cells. In a recent study, we used primary cultures of representative human pheochromocytoma/paraganglioma to establish the cytotoxicity of camptothecin, a prototypical TOP1 inhibitor. We then used a mouse pheochromocytoma cell line (MPC) to further test the effect of camptothecin in conjunction with 5-azacytidine (5-aza), a DNA methylation inhibitor that modulates transcription, and showed that the efficacy of a low concentration of camptothecin is increased by intermittent co-administration of sub-toxic concentrations of 5-aza. The results provide a proof of principle for the use of TOP1 inhibitors against non-dividing pheochromocytoma/paraganglioma cells and suggest novel strategies for enhancing their efficacy and reducing their toxicity by optimizing the combination and timing of their use in conjunction with other drugs.
An animal model of pheochromocytoma and cell culture studies
The lack of sensitive animal models of pheochromocytoma has hindered the study of this tumor and in vivo evaluation of antitumor agents. Recently, we generated two sensitive luciferase models using bioluminescent pheochromocytoma cells: an experimental metastasis model to monitor tumor spreading and a subcutaneous model to monitor tumor growth and spontaneous metastasis. The models offer a platform for sensitive, non-invasive, and real-time monitoring of PHEO primary growth and metastatic burden to follow the course of tumor progression and for testing relevant antitumor treatments in metastatic pheochromocytoma. Currently, we are testing several new drugs in our laboratory on this animal model.
Additional Funding
- PheoPara Alliance 2013, genetic testing, experimental treatments, ongoing
Publications
- Zhuang Z, Yang C, Lorenzo F, Merino M, Fojo T, Kebebew E, Popovic V, Stratakis CA, Prchal JT, Pacak K. Somatic HIF2Α gain-of-function mutations in paraganglioma with polycythemia. N Engl J Med 2012;367:922-930.
- Jochmanova I, Yang C, Zhuang Z, Pacak K. Hypoxia-inducible factor signaling in pheochromocytoma: turning the rudder in the right direction. J Natl Cancer Inst 2013;105:1270-1283.
- Pacak K, Jochmanova I, Prodanov T, Yang C, Merino MJ, Fojo T, Prchal JT, Tischler AS, Lechan RM, Zhuang Z. New syndrome of paraganglioma and somatostatinoma associated wiht polycythemia. J Clin Oncol 2013;31:1690-1698.
- Shankavaram U, Fliedner SM, Elkahloun AG, Barb JJ, Munson PJ, Huynh TT, Matro JC, Turkova H, Linehan WM, Timmers HJ, Tischler AS, Powers JF, de Krijger R, Baysal BE, Takacova M, Pastorekova S, Gius D, Lehnert H, Camphausen K, Pacak K. Genotype and tumor locus determine expression profile of pseudohypoxic pheochromocytomas and paragangliomas. Neoplasia 2013;15:435-447.
- Giubellino A, Sourbier C, Lee MJ, Scroggins B, Bullova P, Landau M, Ying W, Neckers L, Trepel JB, Pacak K. Targeting heat shock protein 90 for the treatment of malignant pheochromocytoma. PLoS One 2013;8:e56083.
Collaborators
- Clara C. Chen, MD, Nuclear Medicine Department, Clinical Center, NIH, Bethesda, MD
- Graeme Eisenhofer, PhD, Universität Dresden, Dresden, Germany
- Abdel G. Elkahloun, PhD, Genome Technology Branch, NHGRI, NIH, Bethesda, MD
- Tito Fojo, MD, PhD, Medical Oncology Branch, NCI, Bethesda, MD
- Electron Kebebew, MD, Surgery Branch, NCI, Bethesda, MD
- Ron Lechan, MD, PhD, Tufts Medical Center, Boston, MA
- Jacques Lenders, MD, Radboud Universiteit, Nijmegen, The Netherlands
- W. Marston Linehan, MD, Urologic Oncology Branch, NCI, Bethesda, MD
- Alexander Ling, MD, Radiology Department, Clinical Center, NIH, Bethesda, MD
- Lani Mercado-Asis, MD, PhD, University of Santo Tomas, Manila, Philippines
- Maria J. Merino, MD, Pathology Department, NCI, Bethesda, MD
- Corina Millo, MD, PET Department, NIH Clinical Center, Bethesda, MD
- Peter J. Munson, PhD, Center for Information Technology, NIH, Bethesda, MD
- Margarita Raygada, PhD, Program in Reproductive and Adult Endocrinology, NICHD, Bethesda, MD
- James C. Reynolds, MD, Nuclear Medicine Department, Clinical Center, NIH, Bethesda, MD
- Constantine A. Stratakis, MD, D(med)Sci, Program in Developmental Endocrinology and Genetics, NICHD, Bethesda, MD
- Henri Timmers, MD, PhD, Radboud Universiteit, Nijmegen, The Netherlands
- Arthur S. Tischler, MD, PhD, New England Medical Center, Boston, MA
- Robert A. Wesley, PhD, Clinical Epidemiology and Biostatistics Service, NIH Clinical Center, Bethesda, MD
- Zhengping Zhuang, MD, PhD, Surgical Neurology Branch, NINDS, Bethesda, MD
Contact
For more information, email karel@mail.nih.gov.