



You are here: Home > Section on Nutrient Control of Gene Expression
Transcriptional and Translational Regulatory Mechanisms in Nutrient Control of Gene Expression

- Alan G. Hinnebusch, PhD, Head, Section on Nutrient Control of Gene Expression
- Yuen Nei Cheung, BS, Predoctoral Fellow
- Wen-Ling Chiu, PhD, Postdoctoral Fellow
- Kamal Dev, PhD, Postdoctoral Fellow
- Jinsheng Dong, PhD, Senior Research Assistant
- Naseem Gaur, PhD, Postdoctoral Fellow
- Daniel Ginsburg, PhD, Postdoctoral Fellow
- Chhabi Govind, PhD, Postdoctoral Fellow
- Cuihua Hu, BA, Special Volunteer
- Iness Jedidi, PhD, Postdoctoral Fellow
- Sebastien Lagiex, PhD, Postdoctoral Fellow
- Pilar Martin-Marcos, PhD, Postdoctoral Fellow
- Eun-Hee Park, PhD, Postdoctoral Fellow
- Hongfang Qiu, PhD, Staff Scientist
- Attiq Rehman, PhD, Postdoctoral Fellow
- Adesh Saini, PhD, Postdoctoral Fellow
- Fan Zhang, MS, Senior Research Assistant
- Fujun Zhou, PhD, Postdoctoral Fellow
We study the regulation of amino acid biosynthetic genes in budding yeast as means of dissecting molecular mechanisms of gene regulation at the translational and transcriptional levels. Transcription of these unlinked genes is coordinately induced by the transcriptional activator GCN4 in response to starvation for any amino acid. Expression of GCN4 is coupled to amino acid levels by a unique translational control mechanism involving four short upstream open reading frames (uORFs) in GCN4 mRNA. Ribosomes translate the 5′-most uORF (uORF1), resume scanning downstream, and under nonstarvation conditions reinitiate translation at uORF2, uORF3, or uORF4 and then dissociate from the mRNA, keeping GCN4 translation repressed. Under starvation conditions, the reinitiating ribosomes bypass uORFs 2–4 and reinitiate at the GCN4 start codon instead. This bypass is triggered by decreased formation of the ternary complex (TC), comprised of initiation factor 2 (eIF2), GTP, and initiator tRNAMet, which binds to the small (40S) ribosomal subunit in assembling the 43S preinitiation complex (PIC). TC abundance is reduced in starved cells by phosphorylation of the alpha subunit of eIF2 (eIF2a) by GCN2, a protein kinase conserved in all eukaryotes, thus converting eIF2 from substrate to inhibitor of its guanine nucleotide exchange factor (GEF), eIF2B. Hence, GCN4 translation is a sensitive in vivo indicator of impaired TC loading on 40S subunits. We previously exploited this fact to isolate mutations in subunits of eIF2B that constitutively derepress GCN4 (Gcd− phenotype) by lowering TC assembly in the absence of eIF2 phosphorylation. More recently, we used the Gcd− selection to identify domains/residues in eIF1, eIF1A, and eIF3 that participate in TC recruitment in vivo. In collaboration with Jon Lorsch's group, we demonstrated that segments/residues in eIF1 and eIF1A implicated in TC recruitment in vivo stimulate this process in a fully reconstituted system in vitro.
We also investigate the roles of various eIFs in scanning the mRNA 5′ untranslated region and accurately identifying the AUG initiation codon. These studies exploit a genetic selection for mutations that elevate initiation at UUG start codons (Sui− phenotype) or suppress this aberrant initiation event (Ssu− phenotype). In this way, we provided the first evidence that eIF1A and the c-subunit of eIF3 make critical contributions to accurate AUG recognition, and we localized these functions to the unstructured N- and C-tails of eIF1A and the N-terminal domain of eIF3c. In collaboration with Lorsch's group, we also provided strong evidence that dissociation of eIF1 from the PIC is a critical step in AUG recognition, indicating that this factor serves as a "gate-keeper" necessary to suppress initiation at non-AUG triplets.
In the arena of transcriptional control, we analyze co-activators required for gene activation by GCN4 to define the molecular program for recruitment of nucleosome-remodeling enzymes and adaptor proteins that remove repressive chromatin structure and recruit TATA-binding protein, other general transcription factors, and RNA polymerase II (Pol II) to promoters. We also analyze association of co-activators with transcribed coding sequences and their roles in transcription elongation, termination, and mRNA export from the nucleus. We recently demonstrated that the histone acetyltransferase complex SAGA is co-transcriptionally recruited to coding sequences in a manner stimulated by phosphorylation of the Pol II C-terminal domain (CTD) by the Kin28 subunit of TFIIH, and that the ensuing acetylation of histone H3 facilitates transcription-associated histone eviction in the coding sequences and enhances the processivity of elongation. More recently, we collaborated with the Rodriguez-Navarro group to show that Sus1, a subunit of both SAGA and the mRNA export complex TREX2, is associated with elongating Pol II and essential mRNA export factors, and that Sus1 promotes transcription elongation. These findings demonstrated that Sus1 acts at the interface between SAGA and TREX2 to coordinate transcription elongation with mRNA export.
Genetic identification of yeast 18S rRNA residues required for recruitment of initiator tRNAMet and AUG selection
We recently applied the Gcd− selection to obtain the first functional evidence implicating 18S rRNA residues of the 40S subunit in TC loading and AUG selection. We found that mutations that perturb the location or identity of the bulge G residue in helix 28 impair TC loading during reinitiation on GCN4 mRNA and, uniquely, increase the bypass of AUG start codons during primary initiation events (leaky scanning). At least one such mutation impairs the rate and stability of TC loading on mutant 40S subunits in vitro. Site-directed mutations in other residues predicted to contact the AUG codon or anticodon loop of tRNAiMet, based on the crystal structures of bacterial 70S ribosome-tRNA complexes, were found to confer dominant Gcd− and recessive-lethal phenotypes. This finding indicates that the tRNA-ribosome interactions present in bacterial elongation complexes are relevant to the binding site of tRNAiMet in eukaryotic PICs. Our results also provide evidence that the flexibility of helix 28, the pivot point for rotation of the "head' domain of the small ribosomal subunit, is a critical parameter for efficient recognition of the AUG start codon during the scanning process (Dong et al., 2008).
Archaeal aIF2B interacts with eukaryotic translation initiation factors eIF2alpha and eIF2Balpha: implications for aIF2B function and eIF2B regulation.
We showed previously that inhibition of eIF2B by phosphorylated eIF2 requires interaction of the “S1” domain of eIF2a with a subcomplex of eIF2B formed by the three regulatory subunits a/GCN3, b/GCD7, and d/GCD2, which blocks the GEF function of the catalytic (e) subunit of eIF2B. The eIF2B regulatory subunits are related in sequence to one another and numerous archaeal proteins, some of which function in methionine salvage or CO2 fixation. Our sequence analyses predicted that members of one phylogenetically distinct group of these archaeal proteins (designated aIF2Bs) are functional homologs of the a, b, and d subunits of eIF2B. In support of our prediction, we showed that three such aIF2B proteins bind to the a subunit of aIF2 from the cognate archaeon, and one aIF2B can also bind to the S1 domain of yeast eIF2a and interact with eIF2Ba/GCN3 in yeast. Mass spectrometry identified several proteins that copurify with aIF2B from the archaeon Thermococcus kodakaraensis, including aIF2a, a sugar-phosphate nucleotidyltransferase with sequence similarity to eIF2Be, and several large subunit (50S) ribosomal proteins. Based on our evidence that aIF2B has functions in common with eIF2B regulatory subunits, the known crystal structure of aIF2B was used to construct a model of the eIF2B regulatory subcomplex, in which evolutionarily conserved regions and sites of regulatory mutations in eIF2B subunits are juxtaposed in a continuous binding surface for phosphorylated eIF2a. Our results provide important structural insights into inhibition of eIF2B by phosphorylated eIF2a, the principal means of down-regulating translation during starvation or stress in eukaryotes.
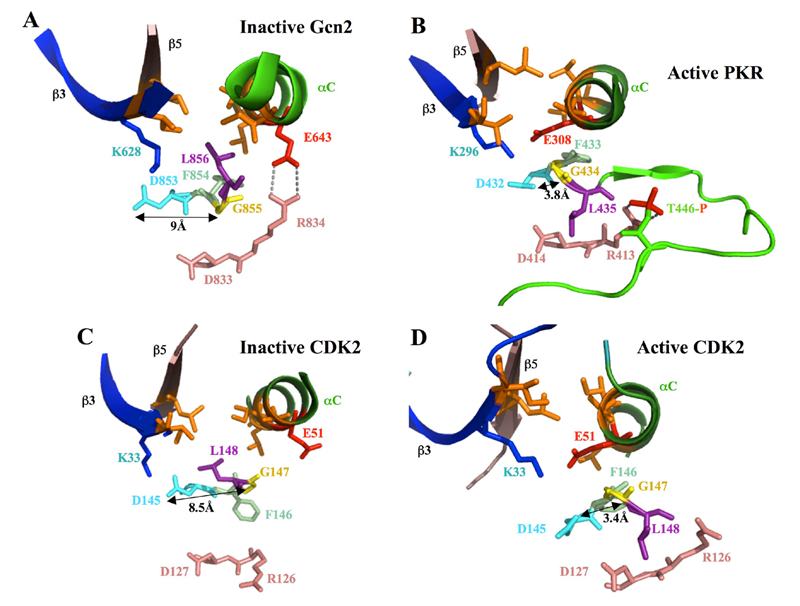
Figure 1. Orientation of the "DFG+1" leucine residue correlates with position of the catalytically important DFG aspartate residue in inactive versus active forms of eIF2a kinases and CDK2
In the structures of inactive forms of Gcn2 (A) and CDK2 (C), interaction of the “DFG+1” Leu residue (L856 or L148, respectively, shown in purple) with hydrophobic residues in β3, β5 and αC (all in orange) correlates with an incorrect position of the Asp (in cyan) of the DFG motif, an important catalytic residue, wherein the ≈9 Å separation between the Asp and Gly (yellow) residues in the DFG motif precludes their H-bonding. In the PKR KD, all but one of the residues corresponding to the L856-hydrophobic network in Gcn2 are perfectly conserved or replaced by a different hydrophobic residue. In the structure of active PKR, considered a model for active Gcn2, the equivalent of Gcn2 L856 (L435) is oriented away from the hydrophobic pocket, adopting the same position as the “DFG+1” Leu residue in the active conformation of CDK2 (panel D). The proposed disruption of the hydrophobic network during kinase activation of Gcn2/PKR (B) and CDK2 (D) could help to achieve the correct position of the DFG Asp and its H bonding to the DFG Gly residue.
A network of hydrophobic residues impeding helix alphaC rotation maintains latency of kinase Gcn2, which phosphorylates the alpha subunit of translation initiation factor 2
The GCN2 kinase domain (KD) is inert and must be activated by binding of uncharged tRNA, which accumulates in starved cells, to the adjacent regulatory domain. We showed previously that Gcn2 latency results from inflexibility of the hinge connecting N- and C-lobes and a partially obstructed ATP-binding site in the KD. We have now demonstrated that a network of hydrophobic interactions centered on Leu-856 also promotes latency by constraining rotation of helix alphaC (αC), depicted in Figure 1A, in a manner relieved by tRNA binding and autophosphorylation of Thr-882 in the KD activation loop. Mutationally disrupting the hydrophobic network constitutively activates eIF2a phosphorylation and bypasses the requirement for tRNA binding and Thr-882 autophosphorylation. In particular, replacing Leu-856 with any non-hydrophobic residue activates Gcn2, while hydrophobic amino acid substitutions maintain latency. We further show that parallel, back-to-back dimerization of the KD is a step in the Gcn2 activation pathway promoted by tRNA binding and autophosphorylation. Remarkably, mutations that disrupt the L856-hydrophobic network or enhance hinge flexibility eliminate the need for the parallel dimer interface, implying that KD dimerization facilitates reorientation of αC and remodeling of the active site for enhanced ATP binding and catalysis. We propose that hinge remodeling, parallel dimerization, and reorientation of αC are mutually reinforcing conformational transitions stimulated by tRNA binding, that are “locked in” by autophosphorylation of T882. Our results on Gcn2 have interesting implications for the latency mechanisms of other important protein kinases, including human eIF2a kinase PKR and human CDK2 (Figure 1), as well as the EGF receptor.
Phosphorylation of the Pol II CTD by KIN28 enhances BUR1/BUR2 recruitment and Ser2 CTD phosphorylation near promoters
The cyclin-dependent kinase BUR1/BUR2 appears to be the yeast ortholog of mammalian P-TEFb, which phosphorylates Ser2 of the Pol II CTD, but the importance of BUR1/BUR2 in CTD phosphorylation was unclear. We have now shown that BUR1/BUR2 is co-transcriptionally recruited to the 5′ end of the Gcn4 target gene ARG1 in a manner stimulated by interaction of the BUR1 C-terminus with CTD repeats phosphorylated on Ser5 by the KIN28 subunit of TFIIH. Remarkably, impairing BUR1/BUR2 function, or removing the CTD-interaction domain in BUR1, reduces Ser2 phosphorylation in bulk Pol II and eliminates the residual Ser2P in cells lacking the major Ser2 CTD kinase CTK1. Impairing BUR1/BUR2 or CTK1 evokes a similar reduction of Ser2P in all elongating Pol II molecules phosphorylated on Ser5 and in elongating Pol II near the ARG1 promoter. By contrast, CTK1 is responsible for the bulk of Ser2P in total Pol II and at promoter-distal sites. Our findings indicate that BUR1/BUR2 is recruited to promoters by Pol II phosphorylated on Ser5 by TFIIH, where it directly phosphorylates the CTD on Ser2 and also stimulates Ser2 phosphorylation by CTK1 (Figure 2). Thus, a cascade of CTD kinases operates to regulate the transition from initiation to elongation and recruit a panoply of factors involved in mRNA processing, elongation, and termination of transcription.
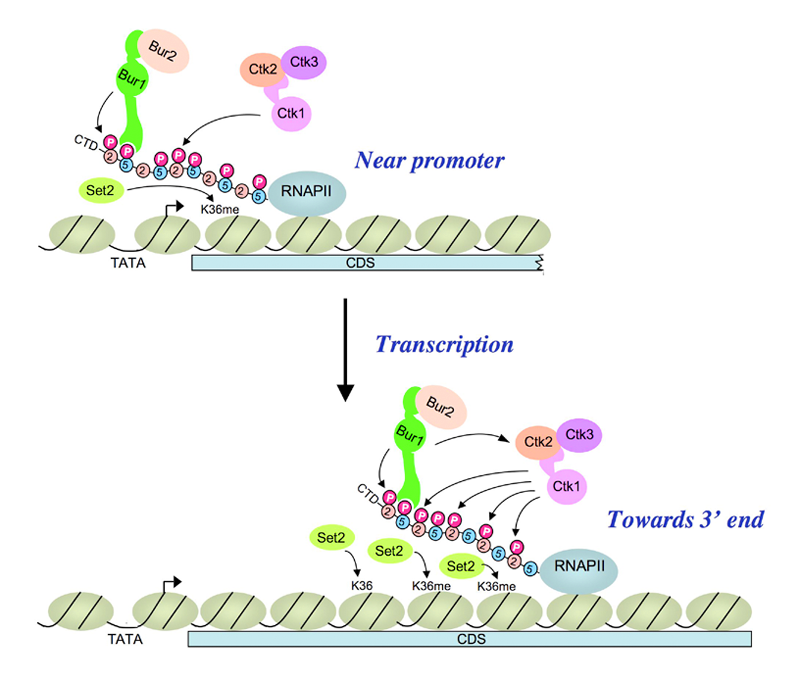
Figure 2. A cascade of cyclin-dependent kinases regulates Pol II CTD phosphorylation in the transcription elongation cycle
(Upper) Kinase BUR1/BUR2 is recruited to Pol II phosphorylated on Ser5 of the heptad repeats in the CTD by KIN28 of TFIIH near the promoter. Recruited BUR1/BUR2 phosphorylates Ser2 of the CTD repeats, enhancing histone H3-Lys36 methylation by SET2. BUR1/BUR2 and CTK1 make similar contributions to phosphorylated Ser2 (Ser2P) at this location. (Lower) As elongation proceeds downstream, kinase CTK1 makes an increasingly larger contribution to Ser2P and attendant H3-Lys36 methylation. Because the occupancies of both kinases remain high at the 3′ end, either CTK1 becomes more active, or BUR1/BUR2 activity declines, as elongation proceeds. Our results suggest that CTK1 function is stimulated by BUR1/BUR2 distal to the promoter.
Disrupting vesicular trafficking at the endosome attenuates transcriptional activation by GCN4
We and others previously identified, on the basis of their sensitivity to sulfometuron methyl (SM) or other inhibitors of amino acid biosynthesis, numerous mutants defective in factors required for translational induction of GCN4 mRNA or lacking co-activators required for transcriptional activation by GCN 4. To identify novel factors involved in general amino acid control, we screened a library of haploid deletion mutants for SM sensitivity (SM). Surprisingly, we identified several SMS/Gcn− strains with deletions of genes involved in vesicular protein trafficking at the late endosome/multivesicular body (MVB). The late endosome/MVB plays a key role in coordinating vesicular transport of proteins among Golgi, vacuole/lysosome, and plasma membrane. These vps mutants are defective in an array of molecules required for producing vesicles with the appropriate cargo proteins or for the tethering and fusion of vesicles at the correct target membranes. We found that GCN4 function is impaired to the greatest extent in class C and D vps mutants, which are defective for various aspects of vesicle fusion at the endosome. Mutations in these factors impair activation of GCN4 target promoters and reduce pre-initiation complex (PIC) assembly at ARG1 without reducing the amount of GCN4 bound to the UASGCRE (upstream activation sequence general control response element) in vivo. We also observed SMS phenotypes for a subset of class E vps mutants, which lack factors needed to sort cargo proteins into intralumenal vesicles (ILV) at the MVB for subsequent transport to the vacuole lumen. The heteromeric protein complexes ESCRT-I, ESCRT-II, and ESCRT-III, which bind ubiquitinated cargo to the MVB outer membrane, carry out the sorting function. The AAA-ATPase VPS4 then recycles E-III and segregates the cargo into ILVs. Class E vps mutants accumulate cargo proteins in aberrant structures lacking ILVs, called class E compartments. The missorted proteins include vacuolar hydrolyases, which are improperly matured but capable of proteolyzing other cargoes that accumulate in the class E compartment. Our detailed analysis of class E mutants indicated that a strong Gcn− phenotype results from protease A–dependent degradation of one or more cargo proteins in the class E compartment rather an inability to transport cargo via ILVs to the vacuole. Combining these results with our finding that defects in endocytosis alone do not confer strong Gcn− phenotypes, we propose that either impaired delivery of MVB cargo originating in the Golgi (class C/D mutants) or rapid proteolysis of the cargo in the aberrant class E compartment (snf7Δ and snf8Δ mutants) is a key condition of MVB dysfunction that leads to a strong reduction in transcriptional activation by GCN4.
Publications
- Dong J, Nanda JS, Rahman H, Pruitt MR, Shin BS, Wong CM, Lorsch JR, Hinnebusch AG. Genetic identification of yeast 18S rRNA residues required forefficient recruitment of initiator tRNAMet and AUG selection. Genes Dev 2008 22:2242-2255.
- Dev K, Santangelo TJ, Rothenburg S, Neculai D, Dey M, Sicheri F, Dever TE, Reeve JN, Hinnebusch AG. Archaeal aIF2B Interacts with Eukaryotic Translation Initiation Factors eIF2alpha and eIF2Balpha: Implications for aIF2B Function and eIF2B Regulation. J Mol Biol 2009 in press.
- Qiu H, Hu C, Hinnebusch AG. Phosphorylation of the Pol II CTD by KIN28 enhances BUR1/BUR2 recruitment and Ser2 CTD phosphorylation near promoters. Mol Cell 2009 33:752-762.
- Sonenberg N, Hinnebusch AG. Regulation of translation initiation in eukaryotes: mechanisms and biological targets. Cell 2009 136:731-745.
- Gárriz A, Qiu H, Dey M, Seo EJ, Dever TE, Hinnebusch AG. A network of hydrophobic residues impeding helix alphaC rotation maintains latency of kinase Gcn2, which phosphorylates the alpha subunit of translation initiation factor 2. Mol Cell Biol 2009 29:1592-1607.
Collaborators
- Thomas Dever, PhD, Program in Cellular Regulation and Metabolism, NICHD, Bethesda, MD
- Jirí Hašek, PhD, Institute of Microbiology, Academy of Sciences of the Czech Republic, Prague, Czech Republic
- Jon Lorsch, PhD, The Johns Hopkins University School of Medicine, Baltimore, MD
- John Reeve, PhD, Ohio State Unversity, Columbus, OH
- Frank Sicheri, PhD, Samuel Lunenfeld Research Institute, Mount Sinai Hospital, Toronto, Canada
- Leos Valášek, PhD, Institute of Microbiology, Academy of Sciences of the Czech Republic, Prague, Czech Republic
Contact
For more information, email hinnebua@mail.nih.gov or visit sncge.nichd.nih.gov.