


You are here: Home > Section on Medical Neuroendocrinology
Diagnosis, Localization, Pathophysiology, and Molecular Biology of Pheochromocytoma and Paraganglioma

- Karel Pacak, MD, PhD, DSc, Head, Section on Medical Neuroendocrinology
- Karen T. Adams, MSc, CRNP, Research Nurse
- Stephanie Fliedner, MS, Visiting Fellow
- Thanh-Truc Huynh, MS, Biologist
- Kathryn King, BS, Postbaccalaureate Fellow
- Lucia Martiniova, MS, Predoctoral Fellow
- Stephen Uyeno, BS, Volunteer
- Samantha Peters, BS, Postbaccalaureate Fellow
We conduct patient-oriented research into the etiology, pathophysiology, genetics, diagnosis, localization, and treatment of pheochromocytoma (PHEO) and paraganglioma (PGL). Projects include both translational research—applying basic science knowledge to clinical diagnosis, pathophysiology, and treatment—and "reverse translation research" by which clinical findings lead to new concepts for pursuit by basic researchers in the laboratory. Our goals are to 1) establish new and improved methods and strategies for novel diagnostic and localization approaches to PHEO and PGL; 2) explain the molecular and cellular basis for varying clinical presentations of PHEO and PGL and establish the pathways of tumorigenesis; 3) search for new molecular and genetic markers for diagnosis and treatment of malignant PHEO and PGL; 4) introduce new therapeutic options for malignant/metastatic PHEO and PGL; and 5) facilitate new and improved collaborations and interdisciplinary studies. To achieve these goals, we base our strategy on multidisciplinary collaborations with investigators from several NIH Institutes and outside medical centers. We link a patient-oriented component with two bench-level components. The patient-oriented component (medical neuroendocrinology) is the driving force for our hypotheses and discoveries. The two bench-level components (tumor pathogenesis and chemistry; biomarkers) emphasize, first, technologies of basic research tailored for pathway and target discovery and, second, the further development of discoveries into clinical applications.
Hereditary PHEOs and PGLs
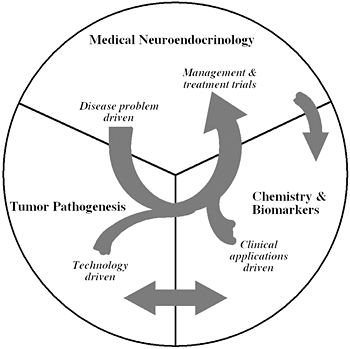
Figure 1
The unique structure of our Section tightly links together a patient-oriented component with two bench-level components. One of the latter two components emphasizes new technologies of basic research tailored for pathway and target discovery; the other focuses on further development of those and other discoveries into clinical applications.
Advances in genetics and the recognition of the high prevalence of PHEO/PGL in certain familial syndromes dictate mandatory tumor screening in patients with those syndromes, irrespective of the presence of classical clinical signs and symptoms. Accumulating data also indicate that many more PHEOs/PGLs result from germline mutations than previously recognized, raising the importance of considering an underlying hereditary condition even in those patients without an obvious family history. To date, mutations in five main genes have been identified as responsible for familial PHEOs/PGLs: 1) the von Hippel-Lindau (VHL) gene in VHL syndrome; 2) the RET gene in multiple endocrine neoplasia type 2 (MEN 2); 3) the neurofibromatosis type 1 (NF-1) gene associated with von Recklinghausen's disease; and 4) mutations in genes encoding mainly the B and D subunits of mitochondrial succinate dehydrogenase (SDHB and SDHD) associated with familial PHEOs/PGLs.
Catecholamine excess and mutations in the genes encoding succinate dehydrogenase subunits (SDHx) are frequently found in patients with PHEOs/PGLs. The genetic screening for PHEO/PGL is rarely done because of time and financial constraints. Therefore, we investigated whether immunohistochemistry of the SDH subunit SDHB could effectively discriminate between SDHx-related and non-SDHx-related PHEOs/PGLs in large prospective and retrospective tumor series. Immunohistochemistry for SDHB was performed on 220 tumors. We investigated two retrospective series of 175 PHEOs/PGLs with known germline mutation status for PHEO/PGL susceptibility genes. Additionally, a prospective series of 45 PHEOs/PGLs were investigated for SDHB immunostaining followed by SDHB, SDHC, and SDHD mutation testing. SDHB protein expression was absent from all 102 PHEOs/PGLs with an SDHB, SDHC, or SDHD mutation, but was present in all 65 PHEOs/PGLs related to MEN 2, VHL syndrome, and NF1. There were 47 out of 53 PHEOs/PGLs with no syndromic germline mutation that showed SDHB expression. The sensitivity and specificity of the SDHB immunohistochemistry to detect the presence of an SDHx mutation in the prospective series were 100% and 84%, respectively.
In another study we assessed the genetic background and clinical characteristics of metastatic PHEO and PGL from a primary tumor found in childhood and adolescence. Forty-five cases of PHEO/PGL in children and adolescents in the age range from 3 to 19 were included. SDHB mutations that included deletions were present in 23 patients (51.1%), VHL in 6 (13.3%), SDHD in 4 (8.9%), NF1 in 2 (4.4%), while 10 patients (22.2%) had sporadic PHEO or PGL. Among patients with SDHB mutations, 22 out of 23 (86.9%) developed metastatic disease. Overall, SDHB mutations were found in 20 out of 28 (71.4%) patients with metastatic disease. Metastatic PHEO or PGL that developed from the primary tumor in childhood and adolescence is most commonly associated with SDHB gene mutations. The presence of an SDHB mutation is often seen in patients with no family history. Determining whether a SDHB gene mutation is present must be considered as the first genetic test to be performed in this patients' population.
Imaging of various PHEOs and PGLs
Although [123I]-MIBG has been in clinical use for imaging of PHEOs/PGLs for many years, a large multicenter evaluation of this agent has never been performed. We designed a study to provide prospective confirmation of the performance of [123I]-MIBG scintigraphy for the evaluation of patients with known or suspected primary or metastatic PHEO/PGL. Eighty-one patients with a history of primary or metastatic PHEO/PGL and 69 with suspicion based on symptoms of catecholamine excess, CT/MRI findings, and/or elevated catecholamines or metanephrines underwent whole-body planar and selected SPECT imaging 24 hours following administration of [123I]-MIBG. Images were independently interpreted by three readers blinded to the status of the study participants, with consensus requiring agreement of at least two readers. Final diagnoses were based on histopathology, correlative imaging, catecholamine or metanephrine measurements, and close clinical follow-up. Among 140 patients with definitive biochemical proof or exclusions of the presence of a tumor (91 positive, 49 negative), [123I]-MIBG planar scintigraphy had sensitivity and specificity of 82%. For patients evaluated for suspected disease, sensitivity and specificity were 88% and 84% respectively. For the subpopulations of adrenal PHEO and extra-adrenal (PGL) tumors, sensitivities were 88% and 67%, respectively. This prospective study demonstrated sensitivity of 82-88% and specificity of 82-84% for [123I]-MIBG imaging used in the diagnostic assessment of primary or metastatic PHEO/PGL.
In another study, patients with negative and borderline-abnormal [123I]-MIBG scans were identified and included if they were diagnosed with PHEO/PGL based on histopathological confirmation of resected tumor or compatible clinical, biochemical, and imaging studies (anatomical and nuclear imaging studies other than [123I]-MIBG). A false-negative [123I]-MIBG scan was defined as absence of uptake corresponding to a lesion proven to be PHEO/PGL by pathology or, when pathologic examination was not available such as in the presence of multiple metastatic lesions, a normal [123I]-MIBG scintigraphy with clinical, biochemical, and imaging findings consistent with PHEO/PGL. We subsequently included 21 patients (6 males, 15 females), seven of whom were referred for evaluation and management of primary tumors preoperatively while 14 other cases were referred due to the development of metastases (n=12), recurrence (n=1), and another primary tumor (n=1) after previous surgery for PHEO/PGL. Eleven cases were due to underlying gene mutation in SDHB and one other case was part of MEN 2 syndrome; nine had benign disease while twelve others had metastatic disease. The results suggest that negative results from [123I]-MIBG scintigraphy are often associated with SDHB-related PHEO or PGL.
Metastatic PHEO and PGL
Expression of carboxypeptidase E (CPE), a prohormone-processing enzyme present in different cancer types was analyzed from data in the Gene Expression Omnibus (GEO) profile database, and experimentally in PHEO/PGL. Microarray data from the GEO profile database indicated that significantly elevated levels of CPE mRNA are found in many non-endocrine cancers: cervical, colon rectal, renal cancers, Ewing sarcomas (bone cancer), and various types of astrocytomas and oligodendrogliomas, but expression of CPE mRNA is virtually absent from their respective counterpart normal tissues. Moreover, there is a good correlation between high CPE mRNA expression and metastasis in cervical cancer, anaplastic oligodendrogliomas, and neoplastic astrocytomas. Elevated CPE mRNA expression was found in neuroendocrine tumors in lung and pituitary adenomas, although the significance is unclear, given that endocrine and neuroendocrine cells normally express CPE. Our studies on PHEOs or PGLs revealed expression of not only wild-type CPE, but of a variant that was correlated with tumor behavior. Extremely high CPE mRNA copy numbers of the variant were found in very large or invasive tumors, which both usually indicate poor prognosis. Thus, all the data suggest that CPE is involved in tumorogenesis and that it may play a role in promoting tumor growth and invasion. CPE could potentially serve as a diagnostic and prognostic biomarker for different cancer types including PHEO/PGL.
Other clinical findings
Biochemical phenotypes of patients with SDHx-related adrenal PHEOS: Ten patients diagnosed with SDHB or SDHD mutations were included in the study, and all underwent adrenalectomy to remove PHEO. Patients with SDHB mutations were found to have elevated plasma or urine metanephrine or normetanephrine or both, while patients with SDHD mutations had elevated plasma norepinephrine only. None of our patients showed elevation in plasma dopamine, DOPAC, urine epinephrine, or dopamine. We concluded that, for SDHx-related adrenal PHEOs, both adrenergic and noradrenergic biochemical phenotypes exist.
Predictive value of tumor size for PHEO metastasis: From a clinical point of view, extra-adrenal tumor location, together with the presence of SDHB mutations, and tumor size have the highest predictive value for the development of metastatic disease. We assessed whether the size of adrenal tumors (usually benign) play a role in the development of metastatic disease regardless of genetic background of these tumors. We found that over 90% of patients presenting with metastatic disease from primary tumor in the adrenal gland had PHEO larger than 5 cm, and most of them (around 60%) developed metastatic disease within five years of the initial diagnosis. Most of these tumors secreted either norepinephrine or norepinephrine together with epinephrine but very rarely only dopamine or epinephrine.
Diagnostic power of the glucagon-stimulation test in PHEO/PGL: Pheochromocytomas can usually be confirmed or excluded using currently available biochemical tests of catecholamine excess. Follow-up tests are, nevertheless, often required to distinguish false-positive from true-positive results. The glucagon stimulation test represents one such test; its diagnostic utility is, however, unclear. The aim of the study was to determine the diagnostic power of the glucagon test to exclude or confirm PHEO/PGL. Glucagon stimulation tests were carried out at three specialist referral centers in 64 patients with PHEO/PGL, 38 patients in whom the tumor was excluded, and in a reference group of 36 healthy volunteers. Plasma concentrations of norepinephrine and epinephrine were measured before and after glucagon administration. Several absolute and relative test criteria were used for calculating diagnostic sensitivity and specificity. Expression of the glucagon receptor was examined in PHEO/PGL tumor tissue from a subset of patients. Irrespective of the various criteria examined, glucagon-provoked increases in plasma catecholamines revealed the presence of the tumor in less than 50% of affected patients. Diagnostic sensitivity was particularly low in patients with PHEO due to VHL syndrome. We concluded that the glucagon stimulation test offers insufficient diagnostic sensitivity for reliable exclusion or confirmation of PHEO/PGL. Because of this and the risk of hypertensive complications, the test should be abandoned in routine clinical practice.
Arterial stiffness in PHEO/PGL patients: The aim of another study was to evaluate arterial stiffness and its modulating factors measured by carotid-femoral pulse wave velocity and central augmentation index in patients with pheochromocytoma before and after surgery. Forty-five patients with PHEO/PGL and 45 healthy controls were investigated using an applanation tonometer (SphygmoCor, AtCor Medical). In addition, 27 patients with PHEO/PGL were studied one year after tumor removal. Pulse wave velocity in PHEO/PGL patients was significantly higher than in controls. Between-group difference in pulse wave velocity remained significant even after adjustment for age, heart rate, fasting plasma glucose, and each of brachial and 24 hr blood pressure parameters. The difference in augmentation index between groups did not reach the statistical significance. Successful tumor removal led to a significant decrease in pulse wave velocity. In conclusion, patients with PHEO/PGL have an increase in pulse wave velocity, which is reversed by the successful tumor removal. Age, mean blood pressure, hs-CRP, and norepinephrine levels are independent predictors of pulse wave velocity.
Survey of anti-hypertensive treatment in PHEO/PGL patients: We recruited 124 PHEO or PGL patients who had been referred to our institute for further management of their tumor for the study (or enrolled prospectively upon admission). The type of anti-hypertensive treatment at the first time when PHEO/PGL was considered was examined. Sixty five per cent (65.2%) of patients who were diagnosed with PHEO/PGL received either no treatment (30%) or inappropriate treatment (35.2%). Only a 34.8% of patients receive accurate treatment. As single therapy, 5.6% of PHEO/PGL patients receive unopposed beta-adrenergic receptor blockade, whereas 16% of patients received alpha-adrenoceptor antagonists in combination with beta-adrenoceptor antagonists or angiotensin-converting enzyme (ACE) inhibitors without a prior period of safe alpha-adrenergic receptor blockade.
Effectiveness of partial adrenalectomy for PHEO: Thirty-six partial adrenalectomies for PHEO were performed in 26 VHL syndrome patients. Twenty-three surgeries were performed using open and 13 using laparoscopic techniques. Prior surgical history was obtained for all patients. At a median followup of 9.25 years (5-46 years), no patient has developed metastatic PHEO. Three patients (11%) developed five local recurrences, treated with surgical extirpation or active surveillance. All recurrences were asymptomatic and detected by radiographic imaging on follow up. Additionally, three of 26 patients (11%) subsequently required partial adrenalectomy for PHEO on the contralateral adrenal gland. In the entire cohort, only three patients became steroid-dependent (11%). Outcomes for partial adrenalectomy in VHL patients with PHEO are encouraging at long-term follow up and should be recommended as a primary surgical approach whenever possible. Adrenal-sparing surgery can obviate the need for steroid replacement in the majority of patients. Local recurrence rates appear to be infrequent and can be managed successfully with subsequent observation or intervention.
Percutaneous radiofrequency ablation of PHEO/PGL metastases: Eight PHEO/PGL metastases (mean size 3.9 cm, range 2.2-7 cm) in six patients aged 31-68 were treated with percutaneous radiofrequency ablation (RFA). All patients underwent pre-ablation adrenergic blockade. Blood pressure measurements and blood samples for plasma catecholamine assay were obtained before, after and at specific intervals during seven of the eight ablation sessions. A predictable temporal relationship was observed between procedural maneuvers, including needle insertion, needle manipulation and application of RFA current, and plasma catecholamines and blood pressure during RFA. With the use of intra-procedural arterial pressure monitoring and peri-procedural adrenergic and catecholamine synthesis blockade, no adverse clinical sequelae due to catecholamine excess and release were observed. Seven of eight metastases underwent complete ablation as defined by tumor non-enhancement on the most recent follow-up CT (mean follow up 10.3 months; range 1-28 months). We concluded that RFA is a safe procedure for treatment of organ or bone lesions in patients with metastatic PHEO/PGL. Patients recover well and quickly, and cardiovascular responses due to catecholamine excess and release can be well controlled by pre-RFA adrenergic and catecholamine synthesis blockade and by careful intra-operative monitoring and treatment.
An animal model of PHEO and cell culture studies
The failure of cytotoxic cancer regimens to cure the most drug-resistant, well-differentiated solid tumors has been attributed to the heterogeneity of cell types that differ in their capacities for growth, differentiation, and metastases. We investigated the ability of LB1, a small molecule inhibitor of serine/threonine protein phosphatase 2A (PP2A), to inhibit a low growth fraction and highly drug-resistant solid neuroendocrine tumor such as metastatic PHEO. Subsequently, we evaluated the increased efficacy of chemotherapy combined with LB1. The effect of LB1 and temozolomide (TMZ), a standard chemotherapeutic agent that alone only transiently suppressed the growth and regression of metastatic PHEO, was evaluated in vitro on PHEO cells and in vivo in a mouse model of metastatic PHEO. We showed that metastatic PHEO, for which there is currently no cure, can be eliminated by combining LB1, thereby inhibiting PP2A, with TMZ. This new treatment approach resulted in long-term, disease-free survival of up to 40% of animals bearing multiple intrahepatic metastases, a disease state from which the majority of patients die. Inhibition of PP2A was associated with prevention of G1/S phase arrest by p53 and of mitotic arrest mediated by polo-like kinase 1 (Plk-1). We propose the elimination of DNA damage–induced defense mechanisms, through transient pharmacologic inhibition of PP2A, as a new approach for enhancing the efficacy of non-specific cancer chemotherapy regimens against a broad spectrum of low growth–fraction tumors very commonly resistant to cytotoxic drugs.
[123I]-MIBG is the most commonly employed treatment for metastatic PHEO/PGL; its success is, however, limited. Its efficacy depends on the [123I]-MIBG concentration achieved within the tumor through its uptake via the membrane norepinephrine transporter and retention in neurosecretory granules. We studied the ability of histone deacetylase (HDAC) inhibitors to enhance [123I]-MIBG uptake by tumors in a mouse metastatic PHEO model. We investigated the in vitro effects of two HDAC inhibitors, romidepsin and trichostatin A, on the uptake of [3H]-norepinephrine and [123I]-MIBG in mouse pheochromocytoma (MPC) cells, and on [18F]-fluorodopamine and [123I]-MIBG uptake in a mouse model of metastatic PHEO. The effects of both inhibitors on norepinephrine transporter activity were assessed in MPC cells by [3H]-norepinephrine and [123I]-MIBG uptake studies with and without the transporter-blocking agent desipramine and the vesicular-blocking agent reserpine. Both HDAC inhibitors enhanced [3H]-norepinephrine, [123I]-MIBG, and [18F]-fluorodopamine uptake through the norepinephrine transporter in MPC cells. In vivo, inhibitor treatment resulted in enhanced uptake of [18F]-fluorodopamine in PHEO liver metastases, as measured by maximal standardized uptake values on PET imaging. Analysis of biodistribution after inhibitor treatment confirmed the PET results. The uptake of [123I]-MIBG was significantly increased in liver metastases (p< 0.05). Therefore, HDAC inhibitor treatment increased radioisotope uptake in MPC cells in vitro and in liver metastases in vivo, through increased norepinephrine transporter activity. HDAC inhibitors may enhance the therapeutic efficacy of [123I]-MIBG treatment in patients with advanced malignant PHEO/PGL.
Additonal Funding
- NICHD
- NICHD
Publications
- Timmers H, Gimenez-Roqueplo AP, Manelli M, Pacak K. Clinical aspects of SDHx-related pheochromocytoma and paraganglioma. Endocr Relat Cancer 2009;16:391-400.
- Lenders JW, Pacak K, Juynh TT, Sharabi Y, Mannelli M, Bratslavsky G, Goldstein DS, Bornstain SR, Eisenhofer G. Low sensitivity of glucagon provocative testing for diagnosis of pheochromocytoma. J Clin Endocrinol Metab. 2010;95:238-245.
- King KS, Whatley MA, Alexopoulos DK, Reynolds JC, Chen CC, Mattox DE, Jacobs S, Pacak K. The use of functional imaging in a patient with head and neck paragangliomas. J Clin Endocrinol Metab. 2010;95:481-482.
- Lodish BM, Adams KT, Huynh TT, Prodanov T, Ling A, Chen C, Shusterman S, Jimenez C, Merino M, Hughes M, Kendall CW, Milosevic D, Singh RJ, Stratakis CA, Pacak K. Succinate dehydrogenase gene mutations are strongly associated with paraganglioma of the organ of Zuckerkandl. Endocr Relat Cancer. 2010;17:581-587.
- Chen H, Sippel RS, O'Dorisi MS, Vinik AI, Lloyd RV, Pacak K. The north american neuroendocrine tumor society consensus guideline for the diagnosis and management of neuroendocrine tumors: pheochromocytoma, paraganglioma, and medullary thyroid cancer. Pancreas. 2010;39:775-783.
- Fliedner SM, Breza J, Kvetnansky R, Powers JF, Tischler AS, Wesley R, Merino M, Lehnert H, Pacak K. Tyrosine hydroxylase, chromogranin A, and steroidogenic acute regulator as markers for successful separation of human adrenal medulla. Cell Tissue Res. 2010;340:607-612.
Collaborators
- Mones Abu-Asab, PhD, Pathology Department, Clinical Center, NIH, Bethesda, MD
- Chris Albanese, PhD, Georgetown University, Washington, DC
- Jorge A. Carrasquillo, MD, Memorial Sloan-Kettering Cancer Center, New York, NY
- Clara C. Chen, MD, Nuclear Medicine Department, Clinical Center, NIH, Bethesda, MD
- Graeme Eisenhofer, PhD, Universität Dresden, Dresden, Germany
- Abdel G. Elkahloun, PhD, Genome Technology Branch, NHGRI, NIH, Bethesda, MD
- Tito Fojo, MD, PhD, Medical Oncology Branch, NCI, Bethesda, MD
- G&B Solutions Inc., McLean, VA
- Jeff Green, MD, PhD, Laboratory of Cancer Biology and Genetics, NCI, Bethesda, MD
- Ioannis Ilias, MD, University of Patras, Patras, Greece
- Electron Kebebew, MD, Surgery Branch, NCI, Bethesda, MD
- Jacques Lenders, MD, St. Radboud University, Nijmegen, The Netherlands
- W. Marston Linehan, MD, Urologic Oncology Branch, NCI, Bethesda, MD
- Alexander Ling, MD, Radiology Department, Clinical Center, NIH, Bethesda, MD
- Maya Lodish, MD, NICHD
- Irina A. Lubensky, MD, Cancer Diagnosis Program, NCI, Bethesda, MD
- Maria J. Merino, MD, Pathology Department, NCI, Bethesda, MD
- John C. Morris, MD, PhD, Metabolism Branch, NCI, Bethesda, MD
- Peter J. Munson, PhD, Center for Information Technology, NIH, Bethesda, MD
- Alan Pang, MD, PhD, Program on Reproductive and Adult Endocrinology, NICHD, Bethesda, MD
- Richard Piekarz, MD, NCI
- Forbes D. Porter, MD, PhD, Program on Developmental Endocrinology and Genetics, NICHD, Bethesda, MD
- Raj K. Puri, MD, PhD, Center for Biologics Evaluation and Research, FDA, Rockville, MD
- Margarita Raygada, PhD, Program on Reproductive and Adult Endocrinology, NICHD, Bethesda, MD
- Douglas Rosing, MD, NHLBI
- James C. Reynolds, MD, Nuclear Medicine Department, Clinical Center, NIH, Bethesda, MD
- Constantine A. Stratakis, MD, D(med)Sci, Program in Developmental Endocrinology and Genetics, NICHD, Bethesda, MD
- David Thomasson, PhD, Radiology Department, Clinical Center, NIH, Bethesda, MD
- Henri Timmers, MD, PhD, St. Radboud University, Nijmegen, The Netherlands
- Arthur S. Tischler, MD, PhD, New England Medical Center, Boston, MA
- Aradhana Venkatesan, MD, Clinical Center, NIH, Bethesda, MD
- Robert A. Wesley, PhD, Clinical Center, NIH, Bethesda, MD
- Jiri Widimsky, MD, PhD, Charles University, Prague, Czech Republic
- Brad Wood, MD, Clinical Center, NIH, Bethesda, MD
- Zhengping Zhuang, MD, PhD, Surgical Neurology Branch, NINDS, NIH, Bethesda, MD
Contact
For more information, email karel@mail.nih.gov.