

You are here: Home > Section on Medical Neuroendocrinology
Diagnosis, Localization, Pathophysiology, and Molecular Biology of Pheochromocytoma and Paraganglioma

- Karel Pacak, MD, PhD, DSc, Head, Section on Medical Neuroendocrinology
- Alessio Giubellino, MD, Senior Research Fellow
- Thanh-Truc Huynh, MS, Biologist
- Karen T. Adams, MSc, CRNP, Research Nurse
- Jan Schovanek, MD, Postdoctoral Visiting Fellow
- Jaydira Del Rivero, MD, Clinical Fellow
- Nikoletta Lendvai, MD, Volunteer
- Ivana Jochmanova, MS, Volunteer
- Petra Bullova, MS, Predoctoral Visiting Fellow
- Joey Matro, MD, Predoctoral Visiting Fellow
- Victoria Martucci, BA, Postbaccalaureate Fellow
We conduct patient-oriented research into the etiology, pathophysiology, genetics, diagnosis, localization, and treatment of pheochromocytoma (PHEO) and paraganglioma (PGL). Projects include both translational research—applying basic science knowledge to clinical diagnosis, pathophysiology, and treatment—and "reverse translation research" by which clinical findings lead to new concepts for pursuit by basic researchers in the laboratory. Our goals are to (i) establish new and improved methods and strategies for novel diagnostic and localization approaches to PHEO and PGL; (ii) explain the molecular and cellular basis for varying clinical presentations of PHEO and PGL and establish the pathways of tumorigenesis; (iii) search for new molecular and genetic markers for diagnosis and treatment of metastatic PHEO and PGL; (iv) introduce new therapeutic options for malignant/metastatic PHEO and PGL; and (v) facilitate new and improved collaborations and interdisciplinary studies. To achieve these goals, we base our strategy on multidisciplinary collaborations with investigators from several NIH Institutes and outside medical centers. We link a patient-oriented component with two bench-level components. The patient-oriented component (medical neuroendocrinology) is the driving force for our hypotheses and discoveries. The two bench-level components (tumor pathogenesis/genetics and chemistry; biomarkers) emphasize, first, technologies of basic research tailored for pathway and target discovery and, second, the further development of discoveries into clinical applications.
Hereditary PHEOs and PGLs
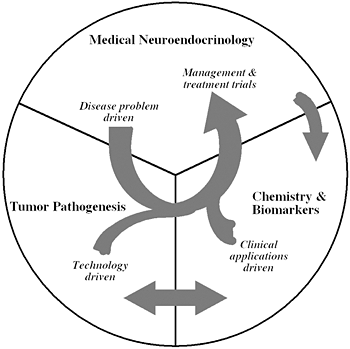
Figure 1
The unique structure of our Section tightly links a patient-oriented component with two bench-level components. One of the latter two components emphasizes new technologies of basic research tailored for pathway and target discovery; the other focuses on further development of those and other discoveries into clinical applications.
MAX has been recently identified as the tenth susceptibility gene for PHEO. However, its clinical relevance was not addressed. An international study, based on an outstanding series of 1694 unrelated patients with PHEO or PGL, ascertained the prevalence of MAX mutations in PHEO patients, extended the spectrum of MAX–related tumors to PGL, revealed contributions of somatic MAX mutations to sporadic disease, and defined an intermediate catecholamine phenotype that may guide testing for the MAX gene in patients with PHEO/PGLs (1). The study also confirmed a preferential paternal mode of transmission with important consequences for genetic counseling. We established that MAX germline mutations are responsible for the disease in 1.12% of cases, similarly to the genes recently described, such as TMEM127, SDHAF2, or SDHA, and now recommend that MAX be considered in the genetic work‐up of affected patients.
Hypoxia-inducible factors (HIFs) are transcription factors controlling energy, iron metabolism, erythropoiesis, and development, and, when dysregulated, contribute to tumorigenesis, cancer progression, and invasion. However, HIFa mutations have not previously been identified in any cancer. We reported two novel somatic gain-of-function HIF2a mutations in two patients, one presenting with paraganglioma and a second with both paraganglioma and somatostatinoma, both associated with polycythemia. Both mutations showed increased HIF2a activity and protein half-life. While germline mutations of HIFa regulators, including VHL, EGLN1, SDHB, SDHC, and SDHD, have been reported in pheochromocytomas/paragangliomas, this was the first report of a somatic gain-of-function mutation in HIF (2).
Mutations of succinate dehydrogenase subunit B (SDHB) play a crucial role in the pathogenesis of the most aggressive and metastatic pheochromocytomas (PHEOs) and paragangliomas (PGLs). Although a variety of missense mutations in the coding sequence of the SDHB gene have been found in PHEOs/PGLs, it was unclear whether these mutations impair mRNA expression, protein stability, subcellular localization, or intrinsic protein function. RT-PCR and Western blot analysis of SDHB mRNA and protein expression from SDHB–related PHEOs/PGLs demonstrated intact mRNA expression but significantly lower protein expression than did non-SDHB PHEOs/PGLS. A pulse chase assay of common SDHB missense mutations in transfected HeLa cell lines demonstrated that the loss of SDHB function was the result of a reduction in mutant protein half-life, whereas co-localization of SDHB with mitochondria and immunoprecipitation with SD–HA demonstrated intact subcellular localization and complex formation. The half-life of the SDHB protein increased after treatment with histone deacetylase inhibitors (HDACi), implicating the protein quality control machinery in the degradation of mutant SDHB proteins. The findings provide the first direct mechanism of functional loss resulting from SDHB mutations and suggest that reducing protein degradation with HDACi may serve as a novel therapeutic paradigm for preventing the development of SDHB–related tumors.
Imaging of various PHEOs and PGLs
We performed a study to compare the sensitivity and specificity of [18F]-FDG PET/CT for tumor localization and staging of PHEO/PGL with conventional imaging by [123I]-MIBG SPECT and CT/MRI. 216 patients (106 males, 110 females) with (suspected) PHEO/PGL were consecutively studied. There were 60 cases of non-metastatic PHEO/PGL, 95 cases of metastatic PHEO/PGL, and 61 PHEO/PGL–negative patients. Besides CT or MRI, patients underwent [18F]-FDG PET/CT and [123I]-MIBG SPECT/CT. For non-metastatic tumors, the sensitivity was 95.7% for CT/MRI, 76.8% for [18F]-FDG (p<0.001 versus CT/MRI), and 77.0% for [123I]-MIBG (p=0.002 versus CT/MRI, not significant versus [18F]-FDG). The specificity was 90.2% for [18F]-FDG, 91.80% for [123I]-MIBG, and 90.2% for CT/MRI. Cut-off values for [18F]-FDG uptake to distinguish between PHEO/PGL and normal adrenal glands were 1.1 (100% sensitivity, 73% specificity) and 4.6 (100% specificity, 82% sensitivity), respectively. [18F]-FDG uptake was higher in SDH- and VHL-related tumors than in MEN2-related tumors. For metastases, sensitivities were 74.4% for CT/MRI, 82.5% for [18F]-FDG (p<0.001 versus [123I]-MIBG), and 50.0% for [123I]-MIBG (p<0.001 versus CT/MRI). For bone metastases, the highest sensitivity was reached by [18F]-FDG: 93.7%, versus 76.7% for CT/MRI (p<0.05) and 61.5% for [123I]-MIBG (p<0.001). Compared with [123I]-MIBG SPECT and CT/MRI, which are all considered the gold standard for PHEO/PGL imaging, metastases were better detected by [18F]-FDG PET. [18F]-FDG PET is highly specific in cases with a biochemically established diagnosis of PHEO/PGL. Quantification of [18F]-FDG uptake distinguishes well between PHEO/PGL and normal adrenal glands and provides important clues for a hereditary syndrome (3).
In another study, we determined the characteristics and outcome of patients with proven PHEO or PGL who had false-negative [123I]-MIBG SPECT. Twenty-one patients with false-negative [123I]-MIBG SPECT (6 males, 15 females), aged 13–55 years, were included. We classified them as non-metastatic or metastatic according to the stage of the disease at the time of false-negative [123I]-MIBG SPECT study, the location and size of the tumor, plasma and urinary catecholamine and metanephrine levels, genetic mutations, and outcome in terms of occurrence and progression of metastases and death. Thirteen patients were evaluated for metastatic tumors while the remaining eight were seen for non-metastatic disease. All primary tumors and multiple metastatic foci did not show avid [123I]-MIBG uptake regardless of the tumor diameter. The majority of patients had extra-adrenal tumors with hypersecretion of normetanephrine or norepinephrine. SDHB mutations were present in 52% (n=11) of cases, RET mutation in 4% (n=1) while the rest were apparently sporadic. Twenty-four percent (n=5) had metastatic disease at initial presentation. Fourteen patients were followed for 3–7 years, of whom 71% (n=10) had metastatic disease and the majority had SDHB mutations. Nine are still alive, while 5 (4 with SDHB) died due to metastatic disease. False-negative [123I]-MIBG SPECT is frequently related to metastatic tumors and usually attributable to SDHB mutations with unfavorable prognosis. We therefore recommend that patients with false-negative [123I]-MIBG SPECT be tested for SDHB mutations and undergo more regular and close follow-up.
Metastatic PHEO and PGL
The wealth of experience from these clinical studies and our previously introduced animal model of metastatic PHEO helped us to (i) further optimize anatomical and functional imaging methods in this mouse model, (ii) increase the uptake of [123I]-MIBG into PHEO tumors in order to improve the efficacy of [131I]-MIBG in the treatment of patients with metastatic PHEO/PGL, and (iii) introduce new pharmacologic treatment options. We found combined use of microCT and [18F]-FDA PET to be the optimal approach for assessing metastatic liver lesions and MRI to provide a more complete assessment of the extent of metastases than does microCT in this mouse model. The histone deacetylase inhibitors romidepsin and trichostatin A were tested as enhancers of [123I]-MIBG, [18F]-FDA, and [3H]-NE uptake by tumors in a mouse model of metastatic PHEO. We found that romidepsin and trichostatin A profoundly increase the expression of the norepinephrine transporter system (NET) and the uptake of [123I]-MIBG, [18F]-FDA, or [3H]-NE in vitro and in vivo. We concluded that histone deacetylase inhibitors may enhance the therapeutic efficacy of [131I]-MIBG treatment in patients with metastatic PHEO/PGL.
In another study, we investigated the ability of LB1, a small-molecule inhibitor of serine/threonine protein phosphatase 2A (PP2A), to inhibit mouse PHEO cells (MPC) in vitro and in vivo in a mouse model of metastatic PHEO alone and in combination with temozolomide (TMZ). When LB1 or TMZ were used separately or in combination in vitro, they only modestly inhibited MPC cell proliferation. However, in vivo, the combination of both drugs markedly reduced the rate of increase in hepatic tumor volume in all 10 animals, with two animals showing no apparent tumor. There was a slight or no inhibitory effect on tumor growth when these drugs were used separately. We also assessed molecular status, including pAkt, pPlk-1, p53, pMDM2, and cell cycle status. Inhibition of PP2A was associated with the prevention of G1/S phase arrest and of mitotic arrest mediated by Plk-1. We proposed that a transient pharmacologic inhibition of PP2A could represent a new approach for enhancing the efficacy of non-specific cancer chemotherapy regimens against a broad spectrum of low growth fraction tumors commonly resistant to cytotoxic drugs.
In collaboration with other groups, we introduced N-terminal truncated carboxypeptidase E (CPE) splice isoform and TNF receptor–associated protein 1 (TRAP-1) as potential new markers and treatment targets (e.g., gamitrinibs inhibit TRAP-1) for malignant or SDHB–related PHEOs/PGLs.
Although PHEOs/PGLs are intensively studied, a highly effective treatment for metastatic PHEO or PGL has not yet been established. Preclinical evaluations of novel therapies for these tumors are thus very much required. Therefore, in another study, we tested the effect of triptolide (TTL), a potent nuclear factor-kappaB (NF-kB) inhibitor, on NET, considered to be the gatekeeper for [131I]-MIBG. We measured changes in the mRNA and protein levels of NET and correlated them with proapoptotic factors and metastasis inhibition. The study was carried out on three different stable pheochromocytoma cell lines. We found that blocking NF-kB with TTL or capsaicin increased both NET mRNA and protein levels. Involvement of NF-kB in the upregulation of NET was verified by mRNA silencing of this site and also by using NF-kB antipeptide. Moreover, MIBG transport was increased in TTL-treated cells, and in vivo treatment with TTL significantly reduced metastatic burden in an animal model of metastatic pheochromocytoma. The study showed, for the first time, how NF-kB inhibitors, by a significant upregulation of NET, could be used successfully in the treatment of metastatic PHEO/PGL to increase the efficacy of [131I]-MIBG and by the induction of apoptosis.
An animal model of PHEO and cell culture studies
The lack of sensitive animal models of PHEO has hindered the study of this tumor and in vivo evaluation of antitumor agents. Recently, we generated two sensitive luciferase models using bioluminescent PHEO cells: an experimental metastasis model to monitor tumor spreading; and a subcutaneous model to monitor tumor growth and spontaneous metastasis. The models offer a platform for sensitive, non-invasive, and real-time monitoring of PHEO primary growth and metastatic burden to follow the course of tumor progression and for testing relevant antitumor treatments in metastatic PHEO.
Additional Funding
- PheoPara Alliance 2012, genetic testing, experimental treatments, ongoing
Publications
- Burnichon N, Cascon A, Schiavi F, Paes Morales N, Comino-Mendez I, Abermil N, Inglada-Perez L, de Cubas AA, Amar L, Barontini MB, Bernaldo de Quiros S, Bertherat J, Bignon YJ, Blok MJ, Bobisse S, Borrego S, Castellano M, Chanson P, Chiara MD, Corssmit EP, Giacche M, de Krijger RR, Ercolino T, Girerd X, Gomez-Garcia EB, Gomez-Grana A, Guilhem I, Hes F, Honrado E, Korpershoek E, Lenders JW, Leton R, Mensenkamp AR, Merlo A, Mori L, Murat A. Pierre P, Plouin PF, Prodanov T, Quesada-Charneco M, Qin N, Rapizzi E, Raymond V, Reisch N, Roncador G, Ruiz-Ferrer M, Schillo F, Stegmann AP, Suarez C, Taschin E, Timmers HJ, Tops C, Urioste M, Beuschlein F, Pacak K, Mannelli M, Dahia PL, Opocher G, Eisenhofer G, Gimenez-Roqueplo AP, Robledo M. MAX mutations cause hereditary and sporadic pheochromocytoma and paraganglioma. Clin Cancer Res 2012;18:2828-2837.
- Zhuang Z, Yang C, Lorenzo F, Merino M, Fojo T, Kebebew E, Popovic V, Stratakis CA, Prchal JT, Pacak K. Somatic HIF2Α gain-of-function mutations in paraganglioma with polycythemia. N Engl J Med 2012;367:922-930.
- Timmers HJLM, Chen CC, Carrasquillo JA, Whatley M, Ling A, Eisenhofer G, King KS, Rao JU, Adams KT, Pacak K. Staging and functional characterization of pheochromocytoma and paraganglioma by 18F-FDG PET. J Nat Cancer Inst 2012;104:700-708.
- Fliedner SM, Kaludericic N, Jiang XS, Hansikova H, Hajkova Z, Sladkova J, Limpuanghthip A, Backlund PS, Wesley R, Martiniova L, Jochmanova I, Lendvai NK, Breza J, Yergey AL, Paolocci N, Tischler AS, Zeman J, Porter FD, Lehnert H, Pacak K. Warburg effect's manifestation in aggressive pheochromocytoma and paragangliomas: insights from a mouse cell model applied to human tumor tissue. PLoS One 2012;7:e40949.
- Pacak K, Sirova M, Giubellino A, Lencesova L, Csaderova L, Laukova M, Hudecova S, Krizanova O. NF-κB inhibition significantly upregulates the norepinephrine transporter system, causes apoptosis in pheochromocytoma cell lines and prevents metastasis in an animal model. Int J Cancer 2012;131:2445-2455.
Collaborators
- Chris Albanese, PhD, Georgetown University, Washington, DC
- Jorge A. Carrasquillo, MD, Memorial Sloan-Kettering Cancer Center, New York, NY
- Clara C. Chen, MD, Nuclear Medicine Department, Clinical Center, NIH, Bethesda, MD
- Peter Choyke, MD, Molecular Imaging Program, NCI, Bethesda, MD
- Graeme Eisenhofer, PhD, Universität Dresden, Dresden, Germany
- Abdel G. Elkahloun, PhD, Genome Technology Branch, NHGRI, NIH, Bethesda, MD
- Tito Fojo, MD, PhD, Medical Oncology Branch, NCI, Bethesda, MD
- G&B Solutions Inc., McLean, VA
- Ioannis Ilias, MD, University of Patras, Patras, Greece
- Electron Kebebew, MD, Surgery Branch, NCI, Bethesda, MD
- Ron Lechan, MD, PhD, Tufts Medical Center, Boston, MA
- Jacques Lenders, MD, St. Radboud University, Nijmegen, The Netherlands
- W. Marston Linehan, MD, Urologic Oncology Branch, NCI, Bethesda, MD
- Alexander Ling, MD, Radiology Department, Clinical Center, NIH, Bethesda, MD
- Maya Lodish, MD, Program on Developmental Endocrinology and Genetics, NICHD, Bethesda, MD
- Y. Peng Loh, PhD, Progam in Develomental Neuroscience, NICHD, Bethesda, MD
- Irina A. Lubensky, MD, Cancer Diagnosis Program, NCI, Bethesda, MD
- Lani Mercado-Asis, MD, PhD, University of Santo Tomas, Manila, Philippines
- Maria J. Merino, MD, Pathology Department, NCI, Bethesda, MD
- Corina Millo, MD, PET Department, NIH Clinical Center, Bethesda, MD
- John C. Morris, MD, PhD, Metabolism Branch, NCI, Bethesda, MD
- Peter J. Munson, PhD, Center for Information Technology, NIH, Bethesda, MD
- Richard Piekarz, MD, Cancer Therapy Evaluation Program, NCI, Rockville, MD
- Forbes D. Porter, MD, PhD, Program in Developmental Endocrinology and Genetics, NICHD, Bethesda, MD
- Raj K. Puri, MD, PhD, Center for Biologics Evaluation and Research, FDA, Rockville, MD
- Margarita Raygada, PhD, Program in Reproductive and Adult Endocrinology, NICHD, Bethesda, MD
- James C. Reynolds, MD, Nuclear Medicine Department, Clinical Center, NIH, Bethesda, MD
- Douglas Rosing, MD, Cardiovascular Branch, NHLBI, Bethesda, MD
- Constantine A. Stratakis, MD, D(med)Sci, Program in Developmental Endocrinology and Genetics, NICHD, Bethesda, MD
- Henri Timmers, MD, PhD, St. Radboud University, Nijmegen, The Netherlands
- Arthur S. Tischler, MD, PhD, New England Medical Center, Boston, MA
- Aradhana Venkatesan, MD, Radiology and Imaging Sciences, NIH Clinical Center, Bethesda, MD
- Robert A. Wesley, PhD, Clinical Epidemiology and Biostatistics Service, NIH Clinical Center, Bethesda, MD
- Jiri Widimsky, MD, PhD, Charles University, Prague, Czech Republic
- Bradford Wood, MD, Center for Interventional Oncology, NIH Clinical Center, Bethesda, MD
- Run Yu, MD, PhD, University of California Los Angeles, Los Angeles
- Jiri Zeman, MD, PhD, Charles University, Prague, Czech Republic
- Zhengping Zhuang, MD, PhD, Surgical Neurology Branch, NINDS, Bethesda, MD
Contact
For more information, email karel@mail.nih.gov.