

You are here: Home > Section on Medical Neuroendocrinology
Diagnosis, Localization, Pathophysiology, and Molecular Biology of Pheochromocytoma and Paraganglioma

- Karel Pacak, MD, PhD, DSc, Head, Section on Medical Neuroendocrinology
- Thanh-Truc Huynh, MS, Biologist
- Karen T. Adams, MSc, CRNP, Research Nurse
- Jaydira Del Rivero, MD, Clinical Fellow
- Ingo Janssen, MD, Postdoctoral Visiting Fellow
- Petra Bullova, MS, Predoctoral Visiting Fellow
- Ivana Jochmanova, MS, Predoctoral Visiting Fellow
- Joan Nambuba, BA, Postbaccalaureate Fellow
- Roland Darr, MD, Volunteer
- Nikoletta Lendvai, MD, Volunteer
We conduct patient-oriented research into the etiology, pathophysiology, genetics, diagnosis, localization, and treatment of pheochromocytoma and paraganglioma. Projects include both translational research—applying basic science knowledge to clinical diagnosis, pathophysiology, and treatment—and “reverse translation research,” by which clinical findings lead to new concepts for pursuit by basic researchers in the laboratory. Our goals are to (i) establish new and improved methods and strategies for novel diagnostic and localization approaches to pheochromocytoma/paraganglioma; (ii) explain the molecular and cellular basis for varying clinical presentations of pheochromocytomas/paragangliomas and establish the pathways of tumorigenesis; (iii) search for new molecular and genetic markers for diagnosis and treatment of metastatic pheochromocytoma/paraganglioma; (iv) introduce new therapeutic options for malignant/metastatic pheochromocytoma/paraganglioma; and (v) facilitate new and improved collaborations and interdisciplinary studies. To achieve these goals, we base our strategy on multidisciplinary collaborations with investigators from several NIH Institutes and outside medical centers. We link a patient-oriented component with two bench-level components. The patient-oriented component (medical neuroendocrinology) is the driving force for our hypotheses and discoveries. The two bench-level components (tumor pathogenesis/genetics and chemistry; biomarkers) emphasize, first, technologies of basic research tailored for pathway and target discovery and, second, the further development of discoveries into clinical applications.
Clinical aspects of pheochromocytoma and paraganglioma
Carotid body paragangliomas (PGLs) are rare tumors that often affect patients with SDHx genetic mutations (the gene encoding succinate dehydrogenase). Despite growing evidence that germ-line genetic mutations alter the aggressiveness of PGLs, treatment decisions are currently based only on clinical symptoms and tumor size in patients with carotid body PGLs. We conducted a retrospective analysis of 34 patients with carotid body PGLs who underwent genetic testing and surgical treatment. Recurrence was defined by the return of locoregional disease and/or development of distant metastases. Clinical characteristics and genetic testing results were analyzed as predictors of patient outcomes. Thirty-four patients underwent 41 primary carotid body PGL resections (median follow-up time of 42 months, range: 1–293). Overall survival was 91.2%. Twelve patients had germ-line mutations in SDHB, 17 in SDHD, and five carried no known mutation. Surgical resection of larger tumors was associated with higher operative complications. Tumor size at resection was significantly smaller in patients with SDHB mutations than in patients with non-SDHB mutations (2.1 vs. 3.3 cm). Mutations in the SDHB gene are associated with worse disease-free survival after resection in patients with carotid body PGLs despite earlier intervention, suggesting that a more aggressive surgical approach is warranted in patients with SDHB mutations.
Aggressive surgical resection with intent to cure and surgical debulking procedures are commonly recommended in patients with metastatic pheochromocytoma (PHEO)/PGL. To date, no data have been published on operative outcomes of patients after surgical resection of metastatic PHEO/PGL to determine whether such an approach is appropriate and what factors may be associated with a favorable outcome. We performed a retrospective analysis of 30 patients with metastatic PHEO/PGL who underwent surgical treatment. Clinical characteristics and genetic factors were analyzed as predictors of biochemical response to surgery. Thirty patients underwent a total of 42 operations, with a median follow-up time of 24 months (range 1 to 114 months). Complete disease resection (R0/R1) was achieved in 18 (42.9%) cases; in 24 cases (57.1%) debulking (R2) procedures were undertaken without intent to cure. Complete biochemical remission was achieved in 10 (23.8%) cases and partial biochemical response was achieved in 23 (54.8%) cases. Patients with disease confined to the abdomen were much more likely to achieve and maintain a biochemical response postoperatively than those with extra-abdominal disease. Debulking operations were significantly less likely to achieve or maintain biochemical palliation, with only one patient maintaining a biochemical response 12 months postoperatively. Patients were less likely to obtain pharmacologic independence after debulking, with only two (8.3%) not requiring pharmacotherapy six months after the intervention. Factors not associated with biochemical response to surgery include sex, family history, SDHB mutation status, systemic therapy, and preoperative biochemical profile. We concluded that, depending on the extent of disease, patients with metastatic PHEO/PGL can benefit from aggressive operative intervention and resection with intent to cure. Debulking procedures are unlikely to achieve clinically significant biochemical response, with any biochemical response achieved being very short-lived.
SDH converts succinate to fumarate, uniquely linking the Krebs cycle with oxidative phosphorylation. SDHx mutations result in the accumulation of succinate associated with various metabolic disturbances and a shift to aerobic glycolysis in tumor tissue. Using mass spectrometry, we measured succinate and fumarate levels in mouse PHEO (MPC) and mouse tumor tissue (MTT) cells and in 10 apparently sporadic, 10 SDHB–, 5 SDHD–, and two neurofibromatosis 1–related PHEOs/PGLs and plasma samples. We found that the succinate-to-fumarate ratio was significantly higher in the SDHB– and SDHD–related PGLs than in apparently sporadic and neurofibromatosis 1–related PHEOs/PGLs. To further support our data, we silenced SDHB expression in MPC and MTT cells and evaluated the succinate and fumarate levels. Compared with control samples, SDHB–silenced MTT cells also showed an increase in the succinate-to-fumarate ratio (MTT cells: 2.45 vs. 7.53), similar to the findings in SDHB–related PGLs. The present findings demonstrate, for the first time, a significantly elevated succinate-to-fumarate ratio in SDHB/D–related PGLs and thus suggest that the ratio may be used as a new metabolic marker for the detection of SDHB/D–related PHEOs/PGLs
Hereditary pheochromocytoma and paraganglioma
Many solid tumors, including PHEO and PGL, are characterized by a (pseudo)hypoxic signature. (Pseudo)hypoxia has been shown to promote both tumor progression and resistance to therapy. The major mediators of the transcriptional hypoxic response are hypoxia-inducible factors (HIFs) (1). High levels of HIFs lead to transcription of hypoxia-responsive genes, which are involved in tumorigenesis. PHEOs and PGLs are catecholamine-producing tumors arising from sympathetic- or parasympathetic-derived chromaffin tissue. In recent years, substantial progress has been made in understanding the metabolic disturbances present in PHEO and PGL, especially as a result of the identification of some disease-susceptibility genes. To date, nineteen PHEO and PGL susceptibility genes have been identified. Based on the main transcription signatures of the mutated genes, pheochromocytomas and paragangliomas were divided into two clusters: pseudohypoxic cluster 1 and pseudohypoxic cluster 2, rich in kinase receptor signaling and protein translation pathways. Although the two clusters appear to show distinct signaling pathways, recent data suggest that both clusters are interconnected by HIF signaling as the important driver in their tumorigenesis, and mutations in most PHEO– and PGL–susceptibility genes seem to affect HIFα regulation and its downstream signaling pathways. HIF signaling appears to play an important role in the development and growth of pheochromocytomas and paraganglioma, which could suggest new therapeutic approaches for the treatment of these tumors
Previously, no HIF2A mutations had been identified in any cancer. First, we reported two novel somatic gain-of-function HIF2A mutations in two patients, one presenting with multiple PGLs and a second with both multiple PGLs and multiple duodenal somatostatinoma, both associated with polycythemia (2). Both mutations showed increased HIF2alpha activity and protein half-life. While germline mutations of HIF2α regulators, including VHL, EGLN1, SDHB, SDHC, and SDHD, had been reported in PHEOs/PGLs, this was the first report of a somatic gain-of-function mutation in HIF2α (2). Subsequently, we investigated two additional unrelated patients and found them to present with the same disease cluster (3). Recently, we described new ocular findings in the patients. Our findings indicate the existence of a new syndrome with multiple PGLs and somatostatinomas associated with polycythemia (Pacak-Zhuang syndrome). The new syndrome results from somatic gain-of-function HIF2A mutations, which cause an upregulation of hypoxia-related genes, including that encoding erythropoietin (EPO) and genes important in cancer biology.
Recently, we investigated additional genetic/pathogenetic factors associated with a new clinical entity in patients presenting with PHEO/PGL and polycythemia. Two patients without hypoxia-inducible factor 2α (HIF2A) mutations, who presented with similar clinical manifestations, were analyzed for other gene mutations, including prolyl hydroxylase (PHD) mutations. We found, for the first time, a germ-line mutation in PHD1 in one patient and a novel germ-line PHD2 mutation in a second patient. Both mutants exhibited reduced protein stability with substantial quantitative protein loss, and thus compromised catalytic activities. Due to the unique association of patients’ polycythemia with borderline or mildly elevated EPO levels, we also performed an in vitro sensitivity assay of erythroid progenitors to EPO and for EPO receptor (EPOR) activity. The results indicated increased EPOR activity and inappropriate hypersensitivity of erythroid progenitors to EPO in the patients. In addition, the study indicates that HIF dysregulation owing to PHD mutations plays an important role in the pathogenesis of these tumors and associated polycythemia. The PHD1 mutation appears to be a new member contributing to the genetic landscape of this novel clinical entity. Our results support the existence of a specific PHD1– and PHD2–associated PHEO/PGL polycythemia disorder.
Imaging of various pheochromocytomas and paragangliomas
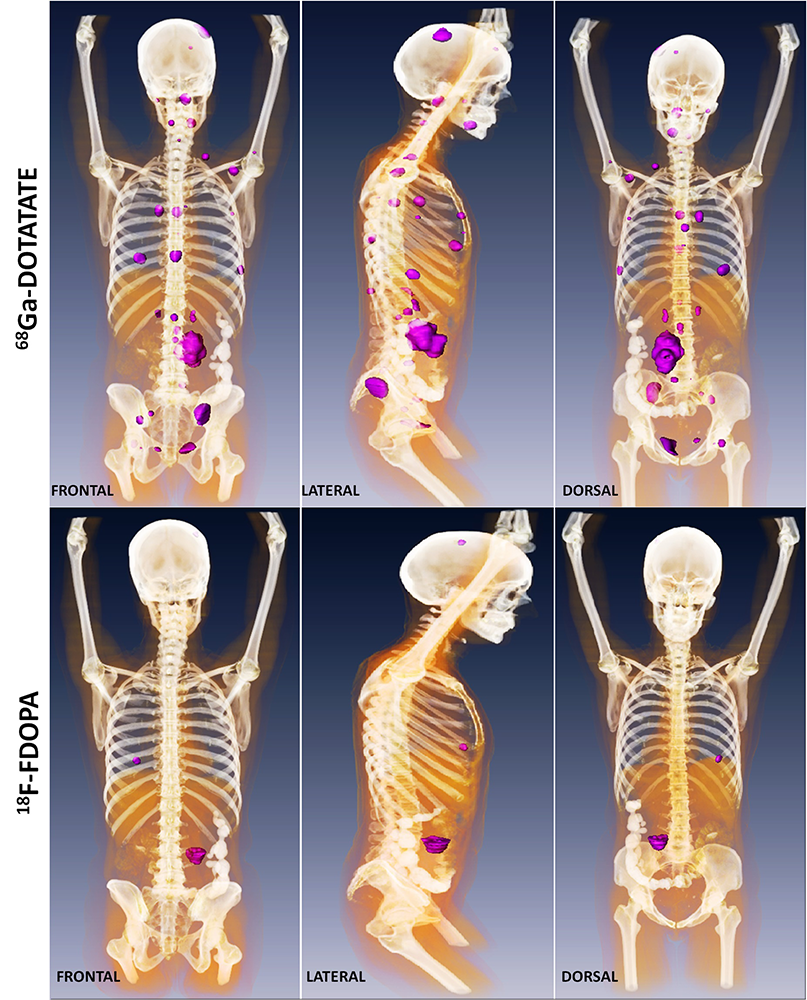
Click image to enlarge.
Metastatic paraganglioma detected by 68Ga-DOTATATE PET/CT and 18F-FDOPA PET/CT
Detection of metastatic paraganglioma with the novel imaging modality 68Ga-DOTATATE PET/CT compared with 18F-FDOPA PET/CT (frontal, lateral, and dorsal views)
Authors: Papadakis GZ, Bagci U, Millo CM, Janssen I, Patronas NJ, Stratakis CA, Pacak K
We studied the feasibility of simultaneous whole-body positron emission tomography/magnetic resonance imaging (PET/MRI) to evaluate patients with PGLs. Fifty-three PGLs or PGL–related lesions from eight patients were evaluated. All patients underwent a single-injection, dual-modality imaging protocol consisting of a PET/CT and a subsequent PET/MRI scan. Four patients were evaluated with 18F-FDG, two with 18F-FDOPA, and two with 18F-FDA. PET/MRI data were acquired using a hybrid whole-body 3-tesla integrated PET/MRI scanner. PET and MRI data (Dixon sequence for attenuation correction and T2-weighted sequences for anatomic allocation) were acquired simultaneously. Imaging workflow and imaging times were documented. PET/MRI and PET/CT data were visually assessed (blindly) with regard to image quality, lesion detection, and anatomic allocation and delineation of the PET findings. With hybrid PET/MRI, we obtained high-quality images in an acceptable acquisition time (median, 31 minutes; range, 25-40 minutes) with good patient compliance. A total of 53 lesions, located in the head and neck area (six lesions), mediastinum (two lesions), abdomen and pelvis (13 lesions), lungs (two lesions), liver (four lesions), and bones (26 lesions), were evaluated. Fifty-one lesions were detected with PET/MRI and confirmed by PET/CT. Two bone lesions (L4 body, 8 mm, and sacrum, 6 mm) were not detectable on an 18F-FDA scan PET/MRI, likely because 18F-FDA was washed out between PET/CT and PET/MRI acquisitions. Coregistered MRI tended to be superior to coregistered CT for head and neck, abdomen, pelvis, and liver lesions for anatomic allocation and delineation. We concluded that clinical PGL evaluation with hybrid PET/MRI is feasible with high-quality image and can be obtained in a reasonable time. It could be particularly beneficial for the pediatric population and for precise lesion definition in the head and neck, abdomen, pelvis, and liver.
Patients with succinate dehydrogenase subunit B (SDHB) mutation–related PHEO/PGL are at high risk for metastatic disease and show worse outcomes compared with other hereditary PHEOs/PGLs. Current therapeutic approaches in these patients are limited, but the best outcomes are based on proper detection of as many lesions as possible. Because PHEOs/PGLs are known to overexpress especially somatostatin receptor 2 (SSTR2), the goal of our study was to assess the eligibility of [68Ga-DOTA0,Tyr3]octreotate (68Ga-DOTATATE) positron emission tomography/computed tomography (PET/CT) in patients with metastatic SDHB–related PHEOs/PGLs. Furthermore, we evaluated its diagnostic value compared with 18F-fluorodopamine (18F-FDA) PET/CT, 18F-L-dihydroxyphenylalanine (18F-FDOPA) PET/CT, 18F-fluoro-2-deoxy-ᴅ-glucose (18F-FDG) PET/CT and 123I-metaiodobenzylguanidine (123I-MIBG) scintigraphy, given that these functional imaging modalities are currently recommended for evaluation and follow-up of these tumors and play an important role in therapeutic plans. 68Ga-DOTATATE PET/CT was performed on 14 patients with SDHB–related metastatic PHEOs/PGLs. All patients also underwent 18F-FDG PET/CT, 13 patients 18F-DOPA PET/CT and 18F-FDA PET/CT. Six patients underwent 123I-MIBG scintigraphy. Sensitivities in the detection of metastatic lesions were compared between all these functional imaging studies. MRI and CT were used as the underlying gold standard. CT or MR identified 213 metastatic lesions. 68Ga-DOTATATE PET/CT demonstrated a lesion-based sensitivity of 97.7 %, detecting 208 lesions. Lesion-based sensitivities for 18F-FDG PET/CT, 18F-FDOPA PET/CT, 18F-FDA PET/CT and 123I-MIBG scintigraphy were 82.2%, 56.3%, 46.7%, and 18.7%, respectively. 68Ga-DOTATATE PET/CT shows significantly higher sensitivity than all other functional imaging modalities in metastatic SDHB–related PHEOs/PGLs. The results suggest that 68Ga-DOTATATE PET/CT may be the preferred imaging modality in metastatic SDHB–related PHEOs/PGLs and may replace currently recommended 18F-FDG PET/CT in the evaluation of these tumors.
Metastatic pheochromocytoma and paraganglioma
Treatment for patients with metastatic PHEO/PGL is lacking. As new PHEO/PGL susceptibility genes are discovered that are associated with the mTOR pathway, treatment targets focusing on this pathway are being intensively explored. Twenty-one human PHEOs/PGLs were analyzed from two tertiary care centers. Immunohistochemistry (IHC) analysis was performed for phospho-mTOR (pmTOR), phospho-S6K (pS6K), phosphoinositide 3-kinase (PI3K), phospho-4EBP1 (p4EBP1), HIF-1α and MIB-1 (mindbomb E3 ubiquitin protein ligase) in six metastatic SDHB PHEOs/PGLs, 15 nonmetastatic PHEOs/PGLs, (including one TMEM127 PHEO and one nonmetastatic SDHB PGL) and six normal adrenal medullas. The product of the intensity of stain and percentage of cells stained was calculated as an H score. Based on a two-sample t-test and paired t-test, pmTOR and pS6K had significantly higher H scores in nonmetastatic PHEOs/PGLs than in metastatic SDHB PHEOs/PGLs. HIF1α had significantly higher H scores in metastatic SDHB PHEOs/PGLs than did nonmetastatic PHEOs/PGLs and normal adrenal medulla. No difference in H scores was seen with p4EBP1, PI3K, and MIB-1 when comparing metastatic SDHB PHEOs/PGLs and nonmetastatic PHEOs/PGLs. Significantly higher pS6K was seen in normal adrenal medullas than in nonmetastatic PHEOs/PGLs and metastatic SDHB PHEOs/PGLs. The results suggest that the use of mTOR inhibitors alone for metastatic SDHB PHEOs/PGLs may not achieve good therapeutic efficacy in patients.
Drug re-purposing or re-positioning has been expanding over the past few years and is now important for the development of new therapeutic options in oncology. We applied this paradigm in the screening of an approximately 3,800–compound library (including FDA–approved drugs and pharmacologically active compounds) employing a model of metastatic PHEO, the most common tumor of the adrenal medulla in children and adults. The collection of approved drugs was screened in quantitative mode, testing the compounds in compound-titration series (dose-response curves). Analysis of the dose-response screening data facilitated the selection of 50 molecules with potential bioactivity in PHEO cells. The drugs were classified based on molecular/cellular targets and signaling pathways affected, and selected drugs were further validated in a proliferation assay and by flow-cytometric cell-death analysis. Using meta-analysis information from molecular targets of the top drugs identified by our screening with gene expression data from human and murine microarrays, we identified potential drugs to be used as single drugs or in combination. Our study exemplified a promising model for identifying potential drugs from a group of clinically approved compounds that can more rapidly be tested in clinical trials of patients with metastatic PHEO/PGL.
An animal model of pheochromocytoma and cell culture studies
The lack of sensitive animal models of PHEO has hindered the study of this tumor and in vivo evaluation of antitumor agents. Recently, we generated two sensitive luciferase models using bioluminescent PHEO cells: an experimental metastasis model to monitor tumor spreading; and a subcutaneous model to monitor tumor growth and spontaneous metastasis. The models offer a platform for sensitive, non-invasive, and real-time monitoring of PHEO primary growth and metastatic burden to follow the course of tumor progression and for testing relevant antitumor treatments in metastatic PHEO. Currently, we are testing several new drugs in our laboratory on this animal model.
Additional Funding
- PheoPara Alliance 2014, genetic testing, experimental treatments, ongoing
Publications
- Zhuang Z, Yang C, Lorenzo F, Merino M, Fojo T, Kebebew E, Popovic V, Stratakis CA, Prchal JT, Pacak K. Somatic HIF2A gain-of-function mutations in paraganglioma with polycythemia. N Engl J Med 2012;367:922-930.
- Jochmanova I, Yang C, Zhuang Z, Pacak K. Hypoxia-inducible factor signaling in pheochromocytoma: turning the rudder in the right direction. J Natl Cancer Inst 2013;105:1270-1283.
- Pacak K, Jochmanova I, Prodanov T, Yang C, Merino MJ, Fojo T, Prchal JT, Tischler AS, Lechan RM, Zhuang Z. New syndrome of paraganglioma and somatostatinoma associated with polycythemia. J Clin Oncol 2013;31:1690-1698.
- Taïeb D, Kaliski A, Boedeker CC, Martucci V, Fojo T, Adler JR Jr, Pacak K. Current approaches and recent developments in the management of head and neck paragangliomas. Endocr Rev 2014;35:795-819.
- Yang C, Zhuang Z, Fliedner SM, Shankavaram U, Sun MG, Bullova P, Zhu R, Elkahloun AG, Kourlas PJ, Merino M, Kebebew E, Pacak K. Germ-line PHD1 and PHD2 mutations detected in patients with pheochromocytoma/paraganglioma-polycythemia. J Mol Med 2014;E-pub ahead of print.
Collaborators
- Clara C. Chen, MD, Nuclear Medicine Department, Clinical Center, NIH, Bethesda, MD
- Graeme Eisenhofer, PhD, Universität Dresden, Dresden, Germany
- Abdel G. Elkahloun, PhD, Genome Technology Branch, NHGRI, NIH, Bethesda, MD
- Tito Fojo, MD, PhD, Medical Oncology Branch, NCI, Bethesda, MD
- Electron Kebebew, MD, Surgery Branch, NCI, Bethesda, MD
- Ron Lechan, MD, PhD, Tufts Medical Center, Boston, MA
- Jacques Lenders, MD, Radboud Universiteit, Nijmegen, The Netherlands
- W. Marston Linehan, MD, Urologic Oncology Branch, NCI, Bethesda, MD
- Alexander Ling, MD, Radiology Department, Clinical Center, NIH, Bethesda, MD
- Lani Mercado-Asis, MD, PhD, University of Santo Tomas, Manila, Philippines
- Maria J. Merino, MD, Pathology Department, NCI, Bethesda, MD
- Corina Millo, MD, PET Department, Clinical Center, NIH, Bethesda, MD
- Peter J. Munson, PhD, Center for Information Technology, NIH, Bethesda, MD
- Margarita Raygada, PhD, Program in Reproductive and Adult Endocrinology, NICHD, Bethesda, MD
- James C. Reynolds, MD, Nuclear Medicine Department, Clinical Center, NIH, Bethesda, MD
- Constantine A. Stratakis, MD, D(med)Sci, Program in Developmental Endocrinology and Genetics, NICHD, Bethesda, MD
- Henri Timmers, MD, PhD, Radboud Universiteit, Nijmegen, The Netherlands
- Arthur S. Tischler, MD, PhD, New England Medical Center, Boston, MA
- Robert A. Wesley, PhD, Clinical Epidemiology and Biostatistics Service, Clinical Center, NIH, Bethesda, MD
- Zhengping Zhuang, MD, PhD, Surgical Neurology Branch, NINDS, Bethesda, MD
Contact
For more information, email karel@mail.nih.gov.