


You are here: Home > Section on Metabolic Regulation
Signal Transduction in Synaptic Transmission and Plasticity

- Kuo-Ping Huang, PhD, Head, Section on Metabolic Regulation
- Freesia L Huang, PhD, Staff Scientist
- Guoxiang Liu, PhD, Visiting Fellow
We investigate the signal transduction mechanisms involved in synaptic transmission and plasticity. Studies of these neural processes are essential to understanding the complex problems related to cognition and behavioral abnormalities. Our current focus is to study genetically modified mice with deletion of the gene encoding neurogranin (Ng), which is specifically expressed in the brain. Ng is a neural-specific protein, which is normally expressed at a high level in subsets of neurons in the forebrain. The protein has been implicated in the regulation of Ca2+/calmodulin (CaM) –dependent cellular processes important for the enhancement of synaptic transmission and plasticity. In humans, mutation of the Ng gene has been linked to behavioral abnormalities and cognitive deficits. Ng levels in the neuronal soma and dendrites are very high, and t he protein sequesters apoCaM at basal physiological Ca2+. Upon synaptic stimulation, the influxed Ca2+ displaces Ng from the Ng/apoCaM complex to form Ca2+/CaM and free Ng. The buffering of CaM by Ng serves as a mechanism to regulate neuronal free Ca2+ and Ca2+/CaM concentrations. The aim of this project is to define the regulatory functions of Ng in neuronal signaling and to design therapeutic approaches to treat cognitive deficits and behavioral disturbances in humans suffering from mutations in Ng.
Stimulation-mediated translocation of calmodulin and neurogranin from soma to dendrites of mouse hippocampal CA1 pyramidal neurons
Calmodulin (CaM) and neurogranin (Ng) are abundant neuronal proteins in the forebrain, and their interactions have been implicated in the enhancement of synaptic plasticity by increasing neurotransmitter-triggered Ca2+ transients. Deletion of the Ng gene in mice causes deficits in learning of hippocampus- and amygdala-dependent behavioral tasks and in high-frequency stimulation (HFS)–induced LTP. We found the concentration of Ng in the hippocampus to be approximately twice that of CaM, and the latter protein was largely concentrated in the nucleus and much less in the dendrites. The asymmetrical distribution of CaM in hippocampal neurons was in contrast to that of Ng, which was abundantly present in the soma and dendrites. Thus, in the distal dendrites, binding of CaM by a relatively higher concentration of Ng renders very little free CaM available for the activation of CaM-dependent enzymes. We explored the possibility that HFS could trigger the mobilization of CaM from soma to dendrites to activate CaMKII and adenylate cyclases for the maintenance of LTP. Tetanic stimulation (a single train of 1 s, 100 Hz) of the Schaffer-collateral fiber of hippocampal slices caused an increase of CaM in the dendrites. HFS-mediated mobilization of CaM was detected surrounding the stimulating electrode within the stratum radiatum but not near a second control electrode that did not deliver HFS. Both CaM and Ng were found to associate with the dendritic spines near the stimulating electrode. Translocation of CaM from soma to dendrites was inhibited by the NMDA receptor antagonist 2-amino-5-phosphonopentanoic acid and was not observed in slices from Ng knockout (KO) mice, which exhibited deficits in HFS-mediated LTP. These findings suggest that association of CaM and Ng at the stimulated dendritic spines may enhance synaptic efficacy by increasing the Ca2+ transients as predicted by a "mass-action mechanism." The association of CaM and Ng at the stimulated synapses may also serve as synaptic tags for the stimulated dendritic branches.
Reduction of extracellular Ca2+ promotes translocation of calmodulin and neurogranin from soma to dendrites of the hippocampal CA 1 neurons.
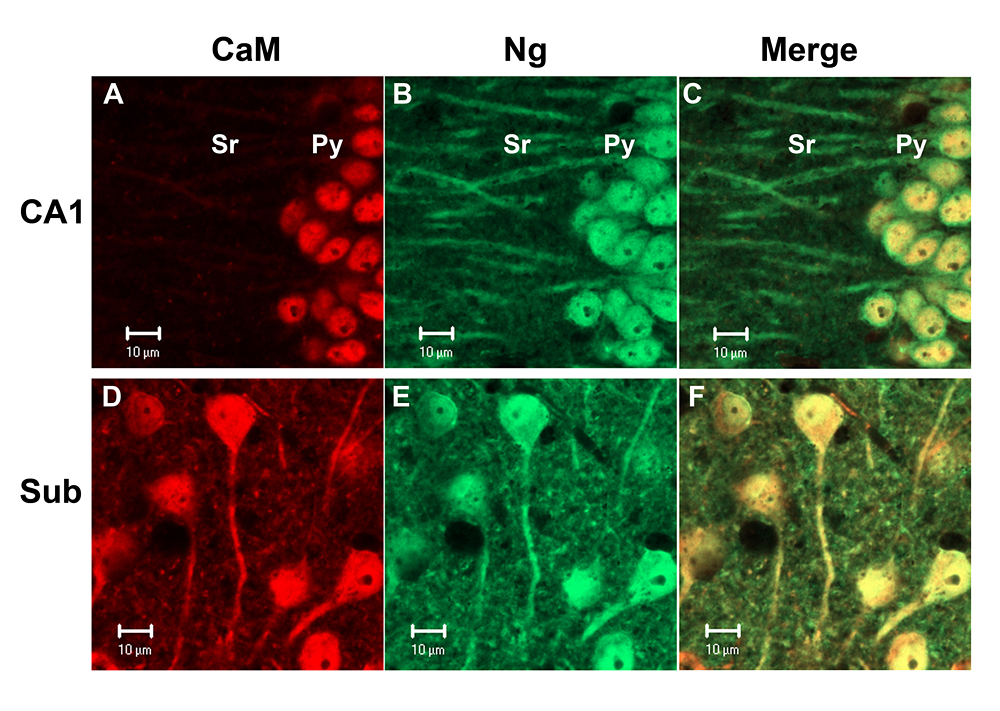
Unique subcellular localization of calmodulin in mouse hippocampal CA1 neurons
Calmodulin (CaM) exhibited strong nuclear localization in mouse hippocampal CA1 pyramidal neurons, but broadly localized in all subcellular compartments in neurons in the subiculum (Sub). In contrast, neurogranin (Ng) was localized in all cellular compartments of these neurons. (click image to enlarge)
Neurogranin (Ng) is a low molecular-weight (7.5 kDa) neuronal protein expressed at a high level in the hippocampus, neocortex, and amygdala. In vitro, Ng binds to apo-CaM, an interaction that is weakened by increasing Ca2+ concentration, its phosphorylation by PKC, and oxidation by nitric oxide and other oxidants. Phosphorylated Ng can stimulate the G protein–coupled phosphoinositide second messengers to trigger the mobilization of Ca2+ from intracellular stores. CaM is believed to be a Ng binding partner in vivo, based on yeast two-hybrid screening. It has been suggested that Ng modulates synaptic responses by buffering and sequestering CaM to regulate the levels of free Ca2+ and Ca2+/CaM complexes. In Ng KOs, neurotransmitter-mediated intracellular Ca2+-transients and down-stream signaling reactions were significantly attenuated compared with wild-type mice. To investigate the dynamics of these two proteins in hippocampal neurons, we tested the effect of extracellular Ca2+ concentrations on the proteins' sub-cellular distribution. Fluorescence immunohistochemical staining of hippocampal slices bathed in Ca2+-containing artificial cerebral spinal fluid (ACSF) revealed that Ng and CaM were co-localized in the soma and dendrites of principle neurons. In the CA1 region the concentrations of CaM and Ng in the soma were greater than those in the dendrites. Surprisingly, majority of the somatic CaM was sequestered in the nucleus. In contrast, Ng was abundantly present in the soma as well as in the dendrites. Changing the bathing fluid from ACSF containing Ca2+ to ACSF that was Ca2+-free (but containing 2.5 mM EGTA) resulted in suppression of synaptic transmission, with a concomitant redistribution of CaM and Ng from soma to dendrites. Confocal Ca2+ imaging showed that reductions of about 15 or 40 nM [Ca2+]i were sufficient to cause half-maximum translocation of Ng and CaM, respectively, from soma to dendrites. Switching the bathing fluid back to Ca2+-containing ACSF restored the synaptic transmission and the original compartmentalization of these two proteins. The hippocampal CA1 pyramidal neurons were more responsive to the Ca2+-sensitive translocation than were their neighboring CA2 and CA3 neurons. The studies illustrated the unique features of hippocampal CA1 neurons in sequestering high concentrations of CaM and Ng in the soma and releasing them to the dendrites when the levels of [Ca2+]i are reduced. The Ca2+-sensitive movement of Ng and CaM between soma and dendrites highlights the unique sensitivity of the CA1 pyramidal neurons to changing [Ca2+]i.
Induction of synaptic facilitation and drug treatment of neurogranin knockout mice
Deletion of Ng in mice caused deficits in cognitive functions and in HFS-induced LTP in the hippocampal CA1 region. Further characterization revealed that such animals also exhibited other behavioral abnormalities, including hyperactivity. The behavioral phenotype likely results from disruption of the Ng-regulated signaling. One of the most prominent roles of Ng is the enhancement of NMDA receptor–mediated Ca2+ transients. Thus, we predict that stimulation of downstream signaling components after Ca2+ influx or increase of the presynaptic transmitter release may rescue the deficits of Ng KO mice. In neurons, stimulation of PKCs is known to enhance transmitter release and facilitate synaptic responses. Short-term treatment of hippocampal slices from Ng KO mice with the PKC-activating phorbol ester phorbol-12, 13-dibutylate (PDBu) caused a persistent synaptic facilitation, lasting for several hours. The PDBu-mediated effects were most prominent among tissue slices from the dorsal hippocampus, which is an area thought to be associated with cognitive functions. Phorbol ester–induced long-term facilitation (LTF) was inhibited by the PKC inhibitor chelerythrine but not by the CaMKII inhibitor KN93, the MEK inhibitor U0126, the protein synthesis inhibitor anisomycin, or the NMDA receptor antagonist AP5. Previously established LTF, however, was only partially inhibited by the same PKC inhibitor, suggesting that activation of PKC is required for the induction of LTF but not for its maintenance. Furthermore, PDBu-mediated LTF occluded the LTP induced by HFS or theta burst stimulation (TBS). On the other hand, TBS-mediated LTP could be further potentiated by PDBu, indicating that activation of PKC induces a new set of plasticity mechanisms in addition to that mediated by TBS.
In conjunction with these in vitro studies, we treated these Ng KO mice with Ritalin®, a psychostimulant drug known to increase extracellular neurotransmitters. Four groups of animals, kept in an enriched environment, including control and drug-treated wild-type and Ng KO mice, were injected with Ritalin® (10 mg/kg/day, i.p.) for three weeks and, afterward, subjected to behavioral tests. The drug-treated Ng KO mice exhibited improvement in their cognitive functions as evidenced by a reduction of the latency time to locate the hidden platform in the water maze and an increase in the freezing time after fear-conditioning. Ritalin® also appeared to reduce the hyperactivity of the Ng KO mice in the open field and an increase in the immobility time in the forced-swim chamber. The drug treatment, however, had only a marginal effect on the performance of the wild-type mice. Measurement of LTP induced by HFS (one train of 100 Hz for 1s) in the hippocampal CA1 region in vitro showed a positive effect of the drug on the Ng KO. These results indicate that Ritalin®, a drug commonly used for the treatment of attention-deficit hyperactivity disorder (ADHD), can exert beneficial effects on the Ng KO, as it does for human patients. These studies also suggest that Ng KO mice will be useful for the development of new treatment strategy for certain behavioral deficits related to ADHD.
Contact
For more information, email kphuang@helix.nih.gov or visit neuroscience.nih.gov/Faculty/Profile/kuo-ping-huang.aspx