



You are here: Home > Section on Macromolecular Recognition and Assembly
Intermolecular Forces, Recognition, and Dynamics

- Donald C. Rau, PhD, Head, Section on Macromolecular Recognition and Assembly
- Shakir Muradymov, PhD, Postbaccalaureate Fellow
- Nina Yu Sidorova, PhD, Staff Scientist
- David Thomas, Summer Intern
Our laboratory focuses on the coupling of forces, structure, and dynamics of biologically important assemblies. The next challenge in structural biology is to understand the physics of interactions between molecules. The ability to take advantage of an increasing number of available protein and nucleic acid structures will depend critically on establishing the link between structure and energy. A fundamental and quantitative knowledge of intermolecular forces is essential for (1) understanding the interactions among biologically important macromolecules that control cellular function and (2) rationally designing agents that can effectively compete with the interactions associated with disease. We have shown that measured forces differ from those predicted by current theories, and we have interpreted the observed forces as indicating the dominant contribution of water-structuring energetics. The observation that interacting macromolecules tenaciously retain their hydration waters unless the surfaces are complementary has profound implications for recognition reactions. To investigate the role of water in binding, we measure and correlate changes in binding energies and hydration that accompany recognition reactions of biologically important macromolecules, particularly sequence-specific DNA-protein complexes. We observe a strong correlation between retained water and binding energy—stronger binding means less water retained at the DNA-protein interface.
Direct force measurements
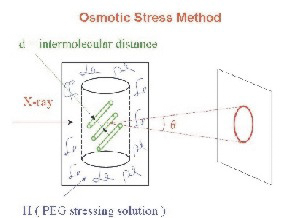
Figure 1. Measurement of intermolecular forces
The osmotic pressure of a polymer as polyethylene glycol (PEG) applies a force on a macroscopic condensed array of ordered biomacromolecules as DNA, lipid bilayers, protein assemblies, or polysaccharides. The consequent distance between macromolecules is measured by X-ray scattering.
The ability to measure directly forces between biopolymers in macroscopic condensed arrays, using the osmotic stress technique illustrated in Figure 1, has greatly changed our understanding of how molecules interact at close spacings—the last 1–1.5 nanometers of separation. The universality of the force characteristics observed for a wide variety of macromolecules, charged or uncharged, including DNA, proteins, lipid bilayers, and carbohydrates, and for the interaction of small solutes and salts with macromolecules has led us to conclude that the energy associated with changes in structuring water between surfaces dominates intermolecular forces. We directly measure forces between biological macromolecules in macroscopic condensed arrays using osmotic stress and X-ray scattering.
DNA-DNA attractions
Dense packaging of DNA in eukaryotic nuclei, sperm nuclei, viruses, and bacteria is necessary for proper cellular functioning. DNA assembly via multivalent ions is a critical testing ground for understanding not only in vivo compaction of DNA but also the physics of interactions between charged molecules. If a sufficient concentration of multivalent ions is present, DNA spontaneously assembles into an ordered array. The helices do not collapse to touching but are rather separated by 0.5–1.5 nm of solvent, depending on the nature of the condensing ion. Attractive and repulsive forces balance at the equilibrium spacing. By combining osmotic stress, pushing experiments with single-molecule magnetic tweezers, and pulling experiments, we were able to separate the attractive and repulsive free energies at the equilibrium spacing for several commonly used condensing agents. The results confirmed our previous hypotheses for hydration forces. There is about a 0.25 nm decay-length exponential repulsive force that is the hydration equivalent of the image charge repulsive force in electrostatics. The hydration atmosphere extending from a solvated surface stabilizes water structuring at the surface. Disruption of the atmosphere by replacing water with another surface lowers hydration energies regardless of the water structuring on the other surface. Repulsion should depend predominately on the water structuring of groups on the DNA surface and perhaps on the mode of binding (phosphate backbone or grooves), but not on the mutual structuring of these groups on apposing helices. The attraction is also characterized by an exponential force but with twice the decay length (about 0.5 nm) of the repulsive force. Attraction results from the direct interaction of surface hydration structures. Perturbations in water structure around one surface resulting from the close presence of another surface can either weaken or strengthen hydration energies depending on the mutual structuring water. We postulated that the attractive force has the same exponential 0.5 nm decay length observed previously for repulsive hydration forces but that the force becomes attractive because of correlations in complementary water structure on apposing helices. By further investigating biogenic alkylamines and arginine peptides of varying lengths, we determined that the magnitude of the attractive force increases with the number of charges, consistent with a constant loss in entropy from correlating a single molecule regardless of charge; the gain in interaction energy increases with the number of charges. The repulsive force is only slightly dependent on the number of charges, as expected, but does depend on the chemical nature of the bound counterion.
We are now investigating the packaging forces of salmon protamine-assembled DNA. Protamines are small, arginine-rich peptides used to package DNA in sperm heads. Protamine–DNA forces resemble polyarginine–DNA interactions. The equilibrium spacing between DNA helices with protamine, however, corresponds to tetra- or penta-arginine, not the 21 arginine charges actually present. We separated protamine–DNA forces into their attractive and repulsive components, finding that the attractive force magnitude itself is very close to that expected for 21 arginine charges whereas the repulsive force is significantly larger than expected from the arginine peptide measurements, suggesting that the neutral amino acids present in protamine result in an increased residual repulsion. We are confirming this hypothesis by measuring DNA force curves with synthetic hexa-arginines that incorporate increasing numbers of neutral amino acids.
We also measured DNA assembly forces in salmon sperm nuclei. The osmotic stress force curves of in vitro, reconstituted DNA–salmon protamine assemblies and in vivo sperm nuclei are virtually identical. Thus, these force measurements have direct biological application. We can use our knowledge of DNA assembly forces to investigate the connection between DNA packaging in sperm heads, DNA damage, and male infertility. There are several reports in the literature correlating human male infertility with protamine abnormalities and deficiencies. Cumulative DNA damage due to reactive oxygen species is a major direct contributor to male infertility, given that DNA repair systems are absent in sperm. We hypothesize that incorrect assembly of DNA by insufficient or altered protamines would result in less tightly packaged DNA in sperm (a larger than normal spacing between helices) and, consequently, a greater potential exposure of DNA to oxidizing free radicals. We can test this hypothesis by directly measuring the distance between helices in sperm of normal and infertile males and of the accumulated DNA damage. We are particularly interested in the effect of incomplete dephosphorylation of protamines on DNA packaging. Protamines are phosphorylated before binding to DNA. Dephosphorylation occurs concurrently with DNA condensation.
Structural and genetic analysis of the X-ray scattering by spores of B. subtilis
Spores of Bacillus species are formed in the process of sporulation; they are metabolically dormant and extremely resistant to a variety of environmental stresses, including heat, radiation, toxic chemicals, and mechanical disruption. The spore coat of the best-studied spore former, Bacillus subtilis, is composed of over 70 proteins unique to spores that assemble into a multi-layered structure, including an outer coat and a lamellar inner coat. The assembly process is complex and not well understood at the molecular level. A number of years ago, X-ray diffraction studies showed that B. subtilis spores contain one or more components that gave two relatively closely spaced X-ray scattering peaks thought to originate from the spore coat. However, the precise identity of the scattering component(s) has not been established, despite the substantial amount of knowledge accumulated recently on the composition and assembly of the B. subtilis spore coat. By examining the X-ray scattering of wild-type B. subtilis spores, as well as spores of a number of isogenic mutants, our combined structural and genetic analyses provided new insight into the structure and assembly of the spore coat. (1) The spore coat alone is responsible for the X-ray scattering peaks observed in dormant B. subtilis spores, as suggested previously from the X-ray scattering of isolated spore coats. (2) A coat protein or proteins whose expression or assembly into the spore coat is either directly or indirectly under the control of the GerE transcription factor is essential for the X-ray scattering peaks. (3) It is likely that these latter GerE–controlled proteins are in the inner spore coat, given that spores from a mutant lacking GerE have no outer coat protein but still give the X-ray scattering peaks. Combined with the knowledge of the spore transcriptome, continued progress in the detailed molecular understanding of spore coat assembly and structure may pave the way for the application of these principles in biotechnology and medicine.
Hydration changes linked to sequence-specific DNA–protein recognition reactions
Our ultimate goal is to apply the lessons from direct force measurements to the recognition reactions that control cellular processes. We have focused on differences in water sequestered by complexes of sequence-specific DNA-binding proteins bound to various DNA sequences, with particular emphasis on correlating binding energy with water incorporated and the energy necessary to remove hydrating water from complexes. We determine differences in sequestered water between complexes through the effect of changing osmotic pressure on binding constants or dissociation rates. Differences in water relate thermodynamics to structure. This focus on water allows us to approach protein-DNA binding from a direction that is very different from standard practice. Indeed, we find that sequestered water correlates quite well with binding energy and is the key difference between specific, non-cognate, and nonspecific complexes.
EcoRV binding to specific and non-specific DNA sequences
We have applied our unique perspective and experimental tools to a controversial type II restriction enzyme: EcoRV. Restriction endonucleases typically show a very high stringency in DNA sequence recognition. There are puzzling results for the relative specific-nonspecific binding constant of EcoRV. Estimates of Ksp-nsp for EcoRV range from 1 to 100, with most estimates less than 10. The possible causes for the inability of EcoRV to distinguish specific from nonspecific sequences are unclear. X-ray crystal structures are available for both cognate and non-cognate EcoRV complexes. The interface of the specific complex is essentially anhydrous with many direct DNA-protein interactions and is very different from the non-cognate complex, which has a large water filled gap at the protein-DNA interface. The substantial difference in structure would suggest a large difference in binding energy.
Unlike our previous observations with EcoRI, we find that the relative specific-nonspecific binding constant for EcoRV is strongly pH-dependent. Specific binding becomes weaker with increasing pH, whereas nonspecific binding is insensitive to pH between 6 and 8. Examination of the X-ray crystal structures, however, does not reveal any histidine–DNA interactions in the specific complex that are absent from the nonspecific complex. The difference in the number of sequestered waters between the specific and nonspecific complexes, however, is insensitive to pH, which would suggest that the solution structures themselves do not change significantly with pH. We would speculate that the X-ray crystal structure of the nonspecific complex does not accurately reflect the solution structure. This may be a general problem for weak complexes; crystal packing forces may be strong enough to distort low-energy nonspecific complexes but not strong specific ones. Our measurements of relative EcoRV specific-nonspecific binding constants indicate significant osmotic pressure dependence. Our estimate of the difference in sequestered water between cognate and nonspecific complexes is about 160 water molecules, a value that correlates well with the difference in structures of the complexes determined by crystallography. This large number of waters further emphasizes that binding specificity within the crowded, high osmotic stress environment of the nucleus likely very differs from that in dilute solution.
Stabilizing labile DNA-protein complexes in polyacrylamide gel using osmolytes
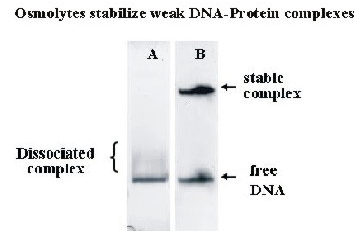
Figure 2. Stabilization of non-cognate DNA-protein complexes in polyacrylamide gels with added osmolytes.
Mixtures of free DNA and nonspecific complexes of the restriction nuclease EcoRI were separated by polyacrylamide gel electrophoresis without (A) and with (B) added osmolyte. The labile complex dissociates quickly in the gel without osmolyte. With 30% triethylene glycol added to the gel, however, the complex is completely stable over the course of the experiment.
In addition to its use as a thermodynamic variable, osmotic stress has several practical applications. Osmotic pressure can greatly stabilize complexes, slowing the dissociation rates by orders of magnitude. We have already taken advantage of this ability to "freeze" reaction mixtures, preventing the dissociation of already formed complexes and the further association of protein and free DNA. We have used osmotic pressure to stabilize labile complexes in polyacrylamide gels. The electrophoretic mobility assay (EMSA) is widely used to monitor the formation of DNA-protein or RNA-protein complexes. The mobility of complexes and free DNA are typically very different in polyacrylamide gel electrophoresis. Many nonspecific DNA-protein complexes, however, are sufficiently weak that they dissociate while migrating, giving smeared bands that are difficult to quantitate. We find that adding osmolytes, in particular triethylene glycol directly to the gel can dramatically stabilize noncognate and nonspecific complexes of the restriction nuclease EcoRI (Figure 2). Extension of this approach to other techniques for separating complex and free components as gel chromatography and capillary electrophoresis is straightforward.
Publications
- Stanley C, Rau DC. Assessing the interaction of urea and protein-stabilizing osmolytes with the nonpolar surface of hydroxypropyl cellulose. Biochemistry 2008 47:6711-6718.
- Todd BA, Parsegian VA, Shirahata A, Thomas TJ, Rau DC. Attractive forces between cation condensed DNA double helices. Biophys J 2008 94:4775-4782.
- Todd B, Stanley C, Sidorova NY, Rau DC. Hydration forces: water and biomolecules. Wiley Encyclopedia of Chemical Biology 2009 1200-1225.
- Todd BA, Rau DC. Interplay of ion binding and attraction in DNA condensed by multivalent cations. Nucleic Acids Res 2008 36:501-510.
- Stanley C, Krueger S, Parsegian VA, Rau DC. Protein structure and hydration probed by SANS and osmotic stress. Biophys J 2008 94:2777-2789.
Collaborators
- Jason DeRouchey, PhD, Program in Physical Biology, NICHD, Bethesda, MD
- William Gelbart, PhD, University of California Los Angeles, Los Angeles, CA
- Susan Krueger, PhD, Center for Neutron Studies, NIST, Gaithersburg, MD
- V. Adrian Parsegian, PhD, Program in Physical Biology, NICHD, Bethesda, MD
- Rudi Podgornik, PhD, Program in Physical Biology, NICHD, Bethesda, MD
- Xiangyun Qiu, PhD, Program in Physical Biology, NICHD, Bethesda, MD
- T.J. Thomas, PhD, University of Medicine and Dentistry of New Jersey–Robert Wood Johnson Medical School, Piscataway, NJ
Contact
For more information, email raud@mail.nih.gov.