

You are here: Home > Section on Medical Neuroendocrinology
Diagnosis, Localization, Pathophysiology, and Molecular Biology of Pheochromocytoma and Paraganglioma

- Karel Pacak, MD, PhD, DSc, Head, Section on Medical Neuroendocrinology
- Alessio Giubellino, MD, Senior Research Fellow
- Hana Turkova, MD, PhD, Visiting Fellow
- Thanh-Truc Huynh, MS, Biologist
- Karen T. Adams, MSc, CRNP, Research Nurse
- Kathryn King, BS, Postbaccalaureate Fellow
- Victoria Martucci, BA, Postbaccalaureate Fellow
- Joey Matro, MD, Predoctoral Visiting Fellow
- Nikoletta Lendvai, MD, Volunteer
- Urvi Shah, MD, Volunteer
We conduct patient-oriented research into the etiology, pathophysiology, genetics, diagnosis, localization, and treatment of pheochromocytoma (PHEO) and paraganglioma (PGL). Projects include both translational research—applying basic science knowledge to clinical diagnosis, pathophysiology, and treatment—and "reverse translation research" by which clinical findings lead to new concepts for pursuit by basic researchers in the laboratory. Our goals are to (i) establish new and improved methods and strategies for novel diagnostic and localization approaches to PHEO and PGL; (ii) explain the molecular and cellular basis for varying clinical presentations of PHEO and PGL and establish the pathways of tumorigenesis; (iii) search for new molecular and genetic markers for diagnosis and treatment of malignant PHEO and PGL; (iv) introduce new therapeutic options for malignant/metastatic PHEO and PGL; and (v) facilitate new and improved collaborations and interdisciplinary studies. To achieve these goals, we base our strategy on multidisciplinary collaborations with investigators from several NIH Institutes and outside medical centers. We link a patient-oriented component with two bench-level components. The patient-oriented component (medical neuroendocrinology) is the driving force for our hypotheses and discoveries. The two bench-level components (tumor pathogenesis and chemistry; biomarkers) emphasize, first, technologies of basic research tailored for pathway and target discovery and, second, the further development of discoveries into clinical applications.
Hereditary PHEOs and PGLs
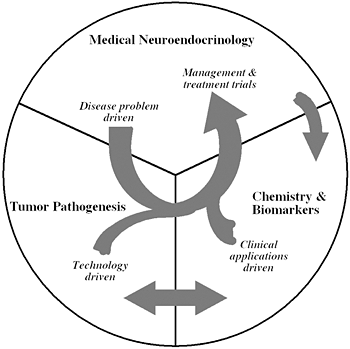
Figure 1
The unique structure of our Section tightly links a patient-oriented component with two bench-level components. One of the latter two components emphasizes new technologies of basic research tailored for pathway and target discovery; the other focuses on further development of those and other discoveries into clinical applications.
We aimed to investigate the rate of SDHB (the gene encoding the succinate dehydrogenase subunit D) mutations in patients with metastatic PHEO/PGL whose initial tumor presentation began in childhood or adolescence. From 2000–2010, we evaluated 263 PHEO/PGL patients. Of those 263 patients, 125 presented with or were found to have metastatic disease; of these 125 patients, 32 presented with a tumor prior to age 20. We performed genetic testing for mutations in the VHL, MEN, and SDHB/C/D genes in patients without previously identified genetic mutations. Of the 32 patients who presented with metastatic disease and with their primary tumor in childhood or adolescence, sequence analysis of germline DNA found SDHB mutations in 23, SDHD mutations in 3, VHL mutations in 2 and absence of a known mutation in 4. The majority (25) of these 32 patients presented with primary tumors in an extra-adrenal location (retroperitoneum and head and neck). We concluded that the majority of patients with metastatic PHEO/PGL who presented with a primary extra-adrenal tumor in childhood or adolescence harbored SDHB mutations. In addition to patients with primary tumors located in the head and neck where SDHD genetic testing is already advised, we recommended that those presenting with metastatic PHEO/PGL in childhood or adolescence undergo SDHB genetic testing.
Eight patients with cardiac PGLs were studied: three had mutations in SDHD, two in SDHB, and one had no SDH mutation; two cases did not undergo genetic testing, although for one patient there was a high likelihood of hereditary PGL because of strong family history. We concluded that SDH gene mutation is most commonly associated with the presence of cardiac PGL.
Diagnosis of PHEOs/PGLs in patients with SDHB and SDHD mutations presents a significant challenge. Currently, measurement of plasma metanephrines (MNs) is the "gold standard" of tumor detection. Previous studies have suggested measuring chromogranin A (CgA) as a secondary diagnostic tool in PHEO/PGL detection. We therefore compared the diagnostic sensitivity of CgA and MNs in SDHB and SDHD patients with proven PHEOs/PGLs. The cohort included 55 patients with SDHB– and 14 patients with SDHD–related proven PHEOs/PGLs. Of the 69 patients, 47 had elevated CgA levels and 48 had elevated plasma MNs. Both CgA and plasma MN levels were elevated in 35, and 60 had either elevated levels of CgA or plasma MNs. In patients with metastatic disease, 73% of SDHB and three out of five SDHD patients had elevated CgA levels, compared with 77% of SDHB and four out of five SDHD patients with elevated plasma MNs. However, CgA or plasma MN levels were elevated in 94% of all SDHB and 100% of SDHD patients with metastatic disease. We found elevated CgA levels in 57% of SDHB and SDHD patients with plasma MN levels within normal range, 41% with plasma MN levels in the "gray zone" between 112ng/mL and 448ng/mL, and 90% with plasma MN levels over four times the upper reference limit. The study thus showed that measurement of CgA can and should be used as sensitive diagnostic marker for patients with hereditary PHEOs/PGLs related to mutations in SDHB or SDHD as complimentary to MNs or alone when MNs are unavailable. We also suggest that CgA measurement be included in the biochemical workup of possible PHEOs/PGLs presented with equivocal or negative catecholamine/metanephrine biochemistry in patients with SDHB or SDHD mutations.
Another study assessed whether measurements of plasma metanephrine, normetanephrine, and methoxytyramine, the O-methylated metabolites of catecholamines, might help distinguish various hereditary forms of the tumor. In 173 patients with PHEO/PGL, including 38 with MEN2 (multiple endocrine neoplasia type 2), 10 with NF1 (neurofibromatosis type 1), 66 with VHL syndrome, and 59 with mutations of the SDHB or SDHD genes, we measured plasma concentrations of O-methylated metabolites by liquid chromatography with electrochemical detection. In contrast to patients with VHL and SDH mutations, all patients with MEN2 and NF1 presented with tumors characterized by increased plasma concentrations of metanephrine (indicating epinephrine production). VHL patients usually showed increases only in normetanephrine (indicating norepinephrine production), whereas additional or solitary increases in methoxytyramine (indicating dopamine production) characterized 70% of patients with SDH mutations. We could distinguish patients with NF1 and MEN2 from those with VHL and SDH mutations in 99% of cases by the combination of normetanephrine and metanephrine measures. Measurements of plasma methoxytyramine discriminated patients with SDH mutations from those with VHL mutations in a further 78% of cases. The distinct patterns of plasma catecholamine O-methylated metabolites in patients with hereditary PHEO/PGL provide an easily used tool to guide cost-effective genotyping of underlying disease-causing mutations.
Imaging of various PHEOs and PGLs
Accurate diagnosis of head and neck PGLs is often complicated by biochemical silence and lack of catecholamine-associated symptoms, making accurate anatomical and functional imaging techniques essential to the diagnostic process. Ten patients (7 SDHD, 3 SDHB), with a total of 26 head and neck PGLs, were evaluated with anatomical and functional imaging. This study compared five different functional imaging techniques in the localization of head and neck PGLs: 18F-fluorodihydroxyphenylalanine (18F-FDOPA) positron emission tomography (PET), 18F-fluorodopamine (18F-FDA) PET/CT, 18F-fluoro-2-deoxy-D-glucose (18F-FDG) PET/CT, 123I-metaiodobenzylguanidine (123I-MIBG) scintigraphy, and 111In-pentetreotide scintigraphy. Prospectively, 18F-FDOPA localized 26/26 lesions in the 10 patients, CT/MRI localized 21/26 lesions, 18F-FDG localized 20/26 lesions, 111In-pentetreotide localized 16/25 lesions, 18F-FDA localized 12/26 lesions, and 123I-MIBG localized 8/26 lesions. Differences in imaging efficacy related to genetic phenotype, even in the present small sample size, included negativity of 18F-FDA and 123I-MIBG in patients with SDHB mutations and accuracy of 18F-FDG in all patients with SDHD mutations, as compared with accuracy of 18F-FDG in only 1 patient with an SDHB mutation. Overall, 18F-FDOPA proved to be the most efficacious functional imaging modality in the localization of SDHx-related head and neck PGLs and may be a potential first line functional imaging agent for the localization of these tumors.
Limited data exist concerning clinical and imaging features that distinguish sporadic from familial PGLs. We studied clinical, genetic (SDHB vs. no SDHx), and imaging (CT, MRI, 18F-FDG PET) features obtained over a decade in 124 PGL patients. Mean age at diagnosis was younger in the SDHB+ group than in the sporadic group (no SDHx) group (28 vs. 39 y, respectively). The rate of supra-diaphragmatic PGLs was higher in the SDHB+ group (17 vs. 5%). Metastasis rates were higher in the SDHB group (79 vs. 48%), as was the existence of metastases or multiple PGLs at presentation (39 vs. 17%). Tumor volumes greater than 250 cc were exclusively observed in SDHB+ patients. On CT, SDHB tumors enhanced more. On 18F-FDG PET, SDHB tumors had higher mean standard uptake values (12.3 vs. 8.0). Clinically, young age, large tumor volume, higher rate of metastatic and multifocal PGLs, higher standardized uptake values (SUV) at FDG PET and increased CT enhancement are observed in SDHB+ PGLs. These findings may warrant genetic screening of patients. Because SDHB patients demonstrate more supra-diaphragmatic lesions, whole body imaging may be of particular value in these patients.
The purpose of another study was to study a series of patients with false-negative 123I-MIBG SPECT (single photon–emission computed tomography) referred to us for evaluation of primary, recurrent, or metastatic PHEO/PGL. We included 21 patients with false-negative 123I-MIBG SPECT (6 males, 15 females), aged 13-55 years (mean 40.9 years). All were proven to have PHEO/PGL by history, biochemical, and imaging studies, or histopathological confirmation of surgically removed tumors. We classified them according to the stage of the disease as non-metastatic, recurrent, or metastatic at the time of false-negative 123I-MIBG study, the location and size of the tumor, plasma and urinary catecholamine and metanephrine levels, genetic mutations, and outcome in terms of occurrence and progression of metastases and death. Eight cases were evaluated for primary tumors, 1 case for a new primary tumor after previous surgery, 1 case for a recurrent, non-metastatic tumor, and 11 for metastatic tumors. All primary tumors and multiple metastatic foci did not show avid 123I-MIBG uptake regardless of the tumor diameter. The majority of patients had extra-adrenal tumors with hypersecretion of normetanephrine and norepinephrine. SDHB mutation was present in 11 cases, RET mutation in 1, and the rest were apparently sporadic. Four had metastatic disease on initial presentation. Fourteen patients were followed up for 3–7 years. Of these, 9 had metastatic disease, while 5 had no metastases. The majority had an SDHB mutation. Nine are still alive, while 5 are deceased due to metastatic disease. A false-negative 123I-MIBG SPECT is frequently related to metastatic tumors and usually due to SDHB mutations with unfavorable prognosis. We therefore recommend that patients with false-negative 123I-MIBG SPECT be tested for SDHB mutations and undergo more regular and close follow-up.
The aim of the final imaging study described in this report study was to establish the diagnostic sensitivity and specificity of 18F-FDG PET/CT for tumor localization and staging of non-metastatic and metastatic PHEO/PGL as compared with conventional anatomical and functional gold-standard imaging by 123I-MIBG SPECT and CT and MRI, respectively. We studied consecutively 216 patients (106 males, 110 females, mean±SD age 45.2±14.9 years) with (suspected) PHEO/PGL. There were 60 cases of non-metastatic PHEO/PGL, 95 cases of metastatic PHEO/PGL and 61 PHEO/PGL-negative patients. Besides CT or MRI, patients underwent 18F-FDG PET/CT and 123I-MIBG SPECT/CT. For non-metastatic tumors, the sensitivity was 96% for CT/MRI, 77% for 18F-FDG and 77% for 123I-MIBG. The specificity was 90% for 18F-FDG, 92% for 123I-MIBG and 90% for CT/MRI. Cut-off values for 18F-FDG uptake to distinguish between PHEO/PGL and normal adrenal glands were 1.1 (100% sensitivity) and 4.6 (100% specificity). 18F-FDG uptake was higher in SDH- and VHL-related tumors than in MEN2-related tumors. For metastases, region-based sensitivities were 74% for CT/MRI, 83% for 18F-FDG, and 50% for 123I-MIBG. For bone metastases, the highest sensitivity was achieved by 18F-FDG: 94%, versus 77% for CT/MRI versus 62% for 123I-MIBG. Compared with 123I-MIBG SPECT, CT, and MRI, all considered gold-standard and the most commonly used imaging methods in these tumors, we found 18F-FDG PET to be the best method for detecting metastases. 18F-FDG PET provides a high specificity in cases of a biochemically established diagnosis of PHEO/PGL. Quantification of 18F-FDG uptake distinguishes well between PHEO/PGL and normal adrenal glands and can provide important clues for a hereditary syndrome underlying PHEO/PGL.
Metastatic PHEO and PGL
This retrospective study focused on clinical, genetic, and histopathologic characteristics of primary metastatic versus primary benign PHEOs. We identified 41 subjects with metastatic PHEO and 108 subjects with apparently benign PHEO. We assessed dimension and biochemical profile of the primary tumor, age at presentation, and time to develop metastases. Subjects with metastatic PHEO presented at a significantly younger age (41.4±14.7 vs. 50.2±13.7 years), with larger primary tumors (8.38±3.27 cm vs. 6.18±2.75 cm) and secreted norepinephrine more frequently (95% vs. 83%) than subjects with apparently benign PHEOs. We found no significant differences in the incidence of genetic mutations in both groups of subjects (26% in the metastatic group and 15% in the benign group). From available histopathologic markers of potential malignancy, only necrosis occurred more frequently in subjects with metastatic PHEO (28% vs. none). The median time to develop metastases was 3.6 years, with the longest interval 24 years. In conclusion, regardless of a genetic background, the size of the primary PHEO and age of first presentation are two independent risk factors associated with the development of metastatic disease.
Another study examined whether measurements of catecholamines and their metabolites would offer utility for diagnosis of PHEO/PGL. Subjects included 365 patients with PHEO/PGL, including 105 with metastases, and a reference population of 846 without PHEO/PGL. We measured 18 catecholamine-related analytes in relation to tumor location and size and mutations of SDHB. Analyses of receiver-operating characteristic curves indicated that plasma methoxytyramine, the O-methylated metabolite of dopamine, provided the most accurate biomarker for discriminating between patients with and without metastases. Plasma methoxytyramine was 4.7-fold higher in patients with than without metastases, a difference that was independent of tumor burden and much larger than those (1.6-1.8 fold) for norepinephrine and its O-methylated metabolite, normetanephrine. Increased plasma methoxytyramine was associated with SDHB mutations and extra-adrenal disease, but was also present in patients with metastases secondary to adrenal tumors or without SDHB mutations. High risk of metastatic disease associated with SDHB mutations reflected large size and extra-adrenal locations of tumors, both independent predictors of malignancy. A plasma methoxytyramine level above 0.2 nmol/L or a tumor diameter above 5 cm indicated an increased likelihood of metastatic disease, particularly when associated with an extra-adrenal location. Plasma methoxytyramine is a novel biomarker for malignant PHEO/PGL that, together with SDHB mutation status and tumor size and location, provides useful information to assess likelihood of malignancy. Biochemical tests to assess the extent of malignant PHEO/PGL, disease progression or response to treatment should include measurements of plasma methoxytyramine.
An animal model of PHEO and cell culture studies
For the rapid advancement and preclinical evaluation of novel therapies for PHEO, it is important to develop reliable and sensitive animal models. To this end, we generated two luciferase models: bioluminescent MTT-luc PHEO cells expressing luciferase were injected into the tail vein for experimental metastasis or implanted subcutaneously to monitor tumor growth. We performed serial in vivo imaging on these animals and ex vivo imaging combined with histopathological analysis at the end of the experiment to confirm and localize metastasis in several organs, as previously detected in vivo. In parallel, we evaluated metastatic burden in the experimental metastasis model using serial MRI. In our experimental metastasis model, localized bioluminescent signals indicating metastasis began to appear, especially in the area of the liver, and were detectable at an early stage of development. Bioluminescent signal correlated with MRI signal, a more traditional imaging technique.
The observation of spontaneous metastasis at distant sites from primary tumor growth at subcutaneous (heterotopic) sites is uncommon, and only a few examples are reported in the scientific literature. Nonetheless, in our subcutaneous model, we were able to detect MTT-luc cells in several organs, especially in the lungs, the organ where subcutaneously implanted MTT-luc cells most consistently metastasized. The animal models offer an opportunity for non-invasive and real-time pre-clinical evaluation of novel therapies and for monitoring development of primary tumors and metastatic burden related to PHEO/PGL.
Publications
- Eisenhofer G, Lenders JW, Siegert G, Bornstein SR, Friberg P, Milosevic D, Mannelli M, Linehan WM, Adams K, Timmers HJ, Pacak K. Plasma methoxytyramine: a novel biomarker of metastatic pheochromocytoma and paraganglioma in relation to established risk factors of tumour size, location and SDHB mutation status. Eur J Cancer 2011;[Epub ahead of print].
- King KS, Prodanov T, Kantorovich V, Fojo T, Hewitt JK, Zacharin M, Wesley R, Lodish M, Raygada M, Gimenez-Roqueplo AP, McCormack S, Eisenhofer G, Milosevic D, Kebebew E, Stratakis CA, Pacak K. Metastatic pheochromocytoma/paraganglioma related to primary tumor development in childhood or adolescence: significant link to SDHB mutations. J Clin Oncol 2011;29:4137-4142.
- Martiniova L, Cleary S, Lai EW, Kiesewetter DO, Seidel J, Dawson LF, Phillips JK, Thomasson D, Chen X, Eisenhofer G, Powers JF, Kvetnansky R, Pacak K. Usefulness of [(18)F]-DA and [(18)F]-DOPA for PET imaging in a mouse model of pheochromocytoma. Nucl Med Biol 2011;[Epub ahead of print].
- King KS, Chen CC, Alexopoulos DK, Whatley MA, Reynolds JC, Patronas N, Ling A, Adams KT, Xekouki P, Lando H, Stratakis CA, Pacak K. Functional imaging of SDHx-related head and neck paragangliomas: comparison of 18F-fluorodihydroxyphenylalanine, 18F-fluorodopamine, 18F-fluoro-2-deoxy-D-glucose PET, 123I-metaiodobenzylguanidine scintigraphy, and 111In-pentetreotide scintigraphy. J Clin Endocrinol Metab 2011;96:2779-2785.
- Eisenhofer G, Lenders JW, Timmers H, Mannelli M, Grebe SK, Hofbauer LC, Bornstein SR, Tiebel O, Adams K, Bratslavsky G, Linehan WM, Pacak K. Measurements of plasma methoxytyramine, normetanephrine, and metanephrine as discriminators of different hereditary forms of pheochromocytoma. Clin Chem 2011;57:411-420.
Collaborators
- Mones Abu-Asab, PhD, Pathology Department, Clinical Center, NIH, Bethesda, MD
- Chris Albanese, PhD, Georgetown University, Washington, DC
- Jorge A. Carrasquillo, MD, Memorial Sloan-Kettering Cancer Center, New York, NY
- Clara C. Chen, MD, Nuclear Medicine Department, Clinical Center, NIH, Bethesda, MD
- Graeme Eisenhofer, PhD, Universität Dresden, Dresden, Germany
- Abdel G. Elkahloun, PhD, Genome Technology Branch, NHGRI, NIH, Bethesda, MD
- Tito Fojo, MD, PhD, Medical Oncology Branch, NCI, Bethesda, MD
- G&B Solutions Inc., McLean, VA
- Jeff Green, MD, PhD, Laboratory of Cancer Biology and Genetics, NCI, Bethesda, MD
- Ioannis Ilias, MD, University of Patras, Patras, Greece
- Electron Kebebew, MD, Surgery Branch, NCI, Bethesda, MD
- Jacques Lenders, MD, St. Radboud University, Nijmegen, The Netherlands
- W. Marston Linehan, MD, Urologic Oncology Branch, NCI, Bethesda, MD
- Alexander Ling, MD, Radiology Department, Clinical Center, NIH, Bethesda, MD
- Maya Lodish, MD, Program on Developmental Endocrinology and Genetics, NICHD, Bethesda, MD
- Irina A. Lubensky, MD, Cancer Diagnosis Program, NCI, Bethesda, MD
- Maria J. Merino, MD, Pathology Department, NCI, Bethesda, MD
- John C. Morris, MD, PhD, Metabolism Branch, NCI, Bethesda, MD
- Peter J. Munson, PhD, Center for Information Technology, NIH, Bethesda, MD
- Richard Piekarz, MD, Cancer Therapy Evaluation Program, NCI, Rockville, MD
- Forbes D. Porter, MD, PhD, Program in Developmental Endocrinology and Genetics, NICHD, Bethesda, MD
- Raj K. Puri, MD, PhD, Center for Biologics Evaluation and Research, FDA, Rockville, MD
- Margarita Raygada, PhD, Program in Reproductive and Adult Endocrinology, NICHD, Bethesda, MD
- Douglas Rosing, MD, Cardiovascular Branch, NHLBI, Bethesda, MD
- James C. Reynolds, MD, Nuclear Medicine Department, Clinical Center, NIH, Bethesda, MD
- Constantine A. Stratakis, MD, D(med)Sci, Program in Developmental Endocrinology and Genetics, NICHD, Bethesda, MD
- Henri Timmers, MD, PhD, St. Radboud University, Nijmegen, The Netherlands
- Arthur S. Tischler, MD, PhD, New England Medical Center, Boston, MA
- Aradhana Venkatesan, MD, Radiology and Imaging Sciences, NIH Clinical Center, Bethesda, MD
- Robert A. Wesley, PhD, Clinical Epidemiology and Biostatistics Service, NIH Clinical Center, Bethesda, MD
- Jiri Widimsky, MD, PhD, Charles University, Prague, Czech Republic
- Bradford Wood, MD, Center for Interventional Oncology, NIH Clinical Center, Bethesda, MD
- Peter Choyke, MD, Molecular Imaging Program, NCI, Bethesda, MD
- Corina Millo, MD, PET Department, NIH Clinical Center, Bethesda, MD
- Ron Lechan, MD, PhD, Tufts Medical Center, Boston
- Lani Mercado-Asis, MD, PhD, University of Santo Tomas, Manila, Philippines
- Run Yu, MD, PhD, University of California Los Angeles, Los Angeles
- Jiri Zeman, MD, PhD, Charles University, Prague, Czech Republic
- Zhengping Zhuang, MD, PhD, Surgical Neurology Branch, NINDS, NIH, Bethesda, MD
Contact
For more information, email karel@mail.nih.gov.