

You are here: Home > Section on Macromolecular Recognition and Assembly
Intermolecular Forces, Recognition, and Dynamics

- Donald C. Rau, PhD, Head, Section on Macromolecular Recognition and Assembly
- Nina Yu Sidorova, PhD, Staff Scientist
- Rene Royal, BA, Postbaccalaureate Fellow
- Andrew Fraser, Summer Intern
Our laboratory focuses on elucidating the coupling of the forces, structure, and dynamics of biologically important assemblies. The next challenge in structural biology is to understand the physics of interactions between molecules in aqueous solution. The ability to take advantage of the increasing number of available protein and nucleic acid structures will depend critically on establishing the link between structures and binding energies. A fundamental and quantitative knowledge of intermolecular forces is necessary for understanding the interactions among biologically important macromolecules that control cellular function and for rationally designing agents that can effectively compete with those interactions associated with disease. Our results have shown that experimentally measured forces at close spacings are very similar for a wide variety of systems but very different from those predicted by current, conventionally accepted theories. We have interpreted the observed forces as indicating the dominating contribution from water-structuring energetics. The observation that interacting macromolecules will tenaciously retain their hydration waters unless the surfaces are complementary has profound implications for recognition reactions. To investigate the role of water in binding, we measure and correlate changes in binding energies and hydration accompanying recognition reactions of biologically important macromolecules, particularly of sequence-specific DNA–protein complexes. We observe a strong correlation between retained water and binding energy; stronger binding means less water retained at the DNA–protein interface.
Direct force measurements
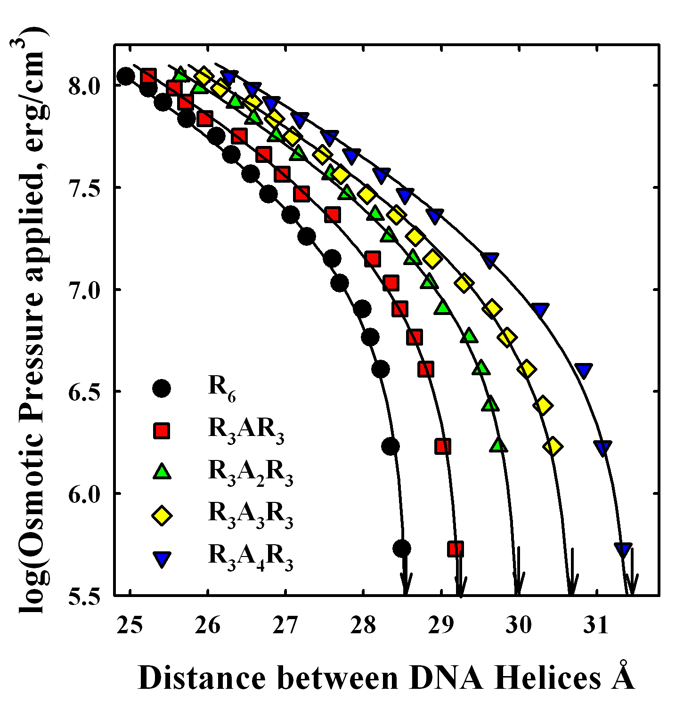
Click image to enlarge.
Figure 1. Alanines inserted into hexa-arginine increase repulsion between DNA helices.
Forces between DNA helices condensed with hexa-arginine peptides with inserted alanines are measured using the osmotic stress technique coupled with x-ray scattering. An increasing number of inserted alanines raises the net repulsion between helices. We conclude that the higher repulsion seen for salmon-protamine–condensed DNA than that expected for 21 arginines is in quantitative agreement with the fraction neutral amino acids present.
DNA assembly and compaction is a necessary feature of cell biology. The DNA in bacteriophages and sperm nuclei is packed densely enough that interhelical spacings and forces can be measured directly. A current goal is to establish a correlation between DNA mispackaging in sperm and male infertility. During vertebrate spermatogenesis, chromatin is dramatically reorganized via histone replacement with protamines to achieve a more compact state for efficient genetic delivery and DNA protection. Dense packaging of DNA in sperm nuclei is considered important to protect DNA against damage by mutagens or oxidative species. Protamines are small, arginine-rich, nuclear proteins that condense the genome into a genetically inactive state. Salmon protamine, for example, is a 32–amino acid peptide with 21 arginines and 11 neutral amino acids. Unlike histone-compacted DNA, the physical properties of reconstituted protamine–DNA assemblies closely resemble those of DNA condensed by well-characterized ions such as divalent manganese, cobalt hexammine, spermidine, spermine, and several arginine oligopeptides. DNA is packaged by protamine to densities within the range seen for DNA condensed with the smaller multivalent ions. More importantly, the forces measured for both reconstituted protamine–DNA arrays and native salmon sperm nuclei show the same characteristics as DNA precipitated by multivalent ions. We hypothesize that male infertility from protamine deficiency and mismodification can be attributed to looser packing of DNA in sperm nuclei than is optimal and greater accessibility to mutagens and oxidizing species.
Upon condensation with the smaller multivalent ions or protamine, the compacted structures have well-defined equilibrium separations of the DNA double helices of 0.7–1.5 nm, depending on the condensing ion. The finite separation of helices indicates a delicate balancing of a short-range repulsive force with a longer-range attraction. The physical origins of the forces acting to compact multivalent ion–DNA assemblies are still being debated. Using combined osmotic stress and single-molecule tweezing experiments, we previously characterized the distance dependence of the two component forces. The attractive force varies with DNA–DNA spacing as a 0.4–0.5 nm decay-length exponential, whereas the repulsive force has a 0.2–0.25 nm decay-length exponential. Using this constraint, we can separate the attractive and repulsive contributions to the force-distance curves. We extracted the amplitudes of the two forces from the force curves for a set of homologous arginine peptides. The magnitude of the attractive force depends on the arginine peptide charge. The repulsive force amplitude, in contrast, is nearly independent of arginine peptide length. The equilibrium spacing between DNA helices in reconstituted salmon protamine–DNA arrays or in salmon sperm nuclei is much larger than expected for 21 arginines. The separation of the forces for salmon protamine–DNA assemblies indicated that the attractive force amplitude with protamine was very close to that expected for 21 arginines, but that the repulsive force had a larger amplitude than expected. This additional repulsion results in the observed lower packaging efficiency of protamine than expected from charge. This is surprising given that cation charge is generally considered to be the dominating determinant of DNA compaction. To determine whether this extra repulsion is a general property of including neutral amino acids in arginine-based peptides or something peculiar to protamine, we investigated the effect on forces of inserting neutral amino acids into model hexa-arginine peptides. Neutral amino acids inserted into hexa-arginine significantly increase the amplitude of short-range repulsive force (Figure 1). We observe a smaller decrease in the amplitude of the attraction. The effect on force amplitudes of incorporating increasing numbers of alanines is additive at least through four, but we observed relatively little difference after inserting alanine, serine, proline, or isoleucine. We find that the increase in the short-range repulsive force for salmon protamine condensed DNA is in good quantitative agreement with its neutral amino acid content. The packing density in salmon sperm nuclei is simply the sum of the arginine and neutral amino acid contributions.
There are several differences between mammalian and piscine protamines. The average arginine fraction of mammalian protamines (50–60%) is significantly smaller than for fish (65–70%). Our results led us to expect that the increase in neutral amino acid content would reduce the net attraction, significantly increasing the equilibrium spacing between helices in mammalian sperm. A looser packaging of DNA would increase accessibility of mutagens and reactive oxidizing species to the DNA. This might explain another difference between mammalian and piscine protamine-DNA packaging. Extensive disulfide bridges between protamines are present in mammalian sperm. Most piscine protamines, on the other hand, do not have cysteines. We hypothesize that the disulfide bridges in mammals are required for tight DNA packaging, overcoming the increased fraction of neutral amino acids.
We also investigated the effect of incorporating a single negative charge—glutamate or phosphorylated serine—into hexa-arginine. The net attraction between DNA helices is substantially weakened, much more than by simply reducing the net charge by one. Indeed, the equilibrium spacing is significantly greater that seen with tri-arginine. The short-range repulsion is dramatically increased. The longer-range attraction is moderately reduced, but is consistent with a net +5 charge.
Incorporation of a negatively charged amino acid into hexa-arginine has a substantial effect on forces, with likely biological implications. The replacement of histones by protamines occurs in several steps. First, histones are replaced by transition proteins, and serine-phosphorylated protamines then replace the transition proteins. It is only after removing the phosphate groups that DNA is tightly packed. Incomplete dephosphorylation has been suggested a cause of increased DNA damage. Our results show that phosphorylation has a much larger effect on weakening the net attraction between helices than simply reducing the protamine charge would suggest.
We recently began to examine mammalian sperm nuclei and are still optimizing techniques. Our preliminary data suggest that, with intact disulfide bridges, stallion sperm DNA is packaged as tightly as in salmon sperm DNA. In agreement with our expectations, the spacing between helices in stallion sperm greatly increases when the disulfide bridges are reduced. We have begun work to find a correlation between DNA–packing densities and infertility.
Hydration changes linked to sequence-specific DNA-protein recognition reactions
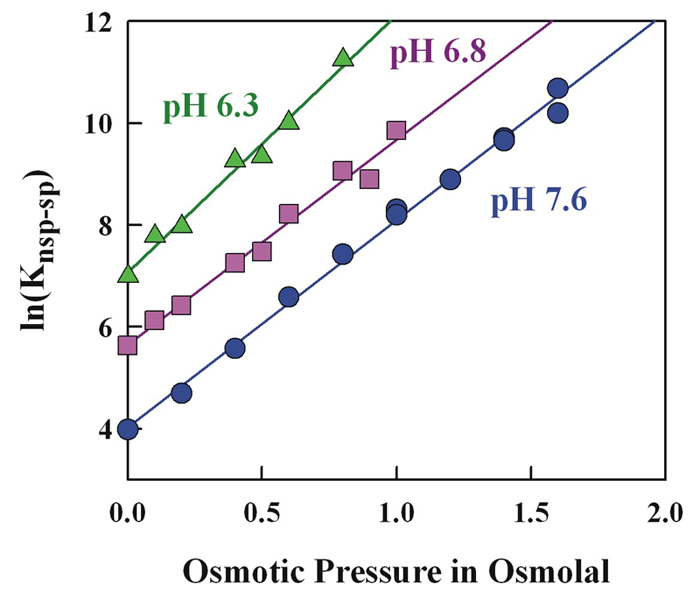
Click image to enlarge.
Figure 2. The large change in relative specific-nonspecific binding constant of EcoRV with DNA with pH is not accompanied by a large change in relative structure.
The osmotic-pressure dependence of the relative specific-nonspecific binding constant of EcoRV gives a difference in water sequestered by the specific and nonspecific protein-DNA complexes. In spite of 20-fold difference in the relative binding constants between pH 6.3 and 7.6, there is very little difference in sequestered water.
The goal of this work is to apply the lessons from direct force measurements to the recognition reactions that control cellular processes. We focused on differences in water sequestered by complexes of sequence-specific DNA–binding proteins bound to different DNA sequences, with particular emphasis on correlating binding energy and water incorporated and on the energy necessary to remove hydrating water from complexes. We determine differences in sequestered water between complexes through the effect of changing osmotic pressure on binding constants or dissociation rates. Differences in water between specific and nonspecific complexes connect thermodynamics and structure. The emphasis on water allows us to approach protein–DNA protein binding from a direction that is different from standard practice. Indeed, we find that sequestered water correlates quite well with binding energy and is the key difference between specific, non-cognate, and nonspecific complexes. We take advantage of the unique experimental tools we have developed to investigate, from a fresh perspective, the coupling of structure, thermodynamics, and function of these complexes.
We are currently investigating DNA complexes of the EcoRV. The literature suggests that, unlike most type II restriction enzymes, EcoRV has low sequence stringency. Using a self-cleavage assay that we developed, we measured EcoRV–DNA competitive binding and evaluated the influence of water activity, pH, and salt concentration on the binding stringency in the absence of divalent ions. We find that the enzyme can readily distinguish specific from nonspecific sequences. The relative specific-nonspecific binding constant increases strongly with increasing osmotic pressure and with decreasing pH. The difference in number of associated waters between specific and nonspecific DNA–EcoRV complexes is consistent with the differences in crystal structures. Despite the large pH dependence of the sequence specificity, the osmotic pressure dependence indicates little change in structure with pH (Figure 2). Importantly, the large osmotic pressure dependence means that measurement of protein–DNA specificity in dilute solution cannot be directly applied to binding in the crowded environment of the cell.
In the absence of divalent ions, EcoRV has unusual kinetics of specific complex formation not observed for EcoRI. A significant fraction of the total enzyme forms enzymatically competent complexes slowly. The slow rate of complex formation could explain the lack of specificity reported by others. This novel result is explained by a very slow transition between two conformations of the free enzyme. We are investigating the pH, salt, osmolyte, and divalent cation dependences of equilibrium distribution of the two structures. The observation of at least two kinetics components in association indicates that EcoRV is an allosteric protein. Allosterism is now recognized as an important concept for DNA–protein complexes, offering an additional level of control over binding and activity. The recognition specificity or activity can be modulated by ligands or proteins that bind to one allosteric conformation in preference to others.
The association and dissociation kinetics of sequence-specific DNA–binding proteins are surprisingly complicated. In association, proteins initially bind nonspecifically and slide along the DNA to either find the specific sequence or dissociate and start the process again. Sliding allows the protein to scan a region of DNA and to locate its recognition sequence faster than solution diffusion would allow. If the recognition sites of transcription factors are normally occluded by nucleosomes, there is a limited time period, during which nucleosomes are transiently displaced, for these factors to find their sites. The dissociation process is just the reverse process. Dissociation and subsequent rebinding to the same DNA is termed hopping or jumping. We have uncovered a novel method to probe hopping kinetics.
The standard method of measuring dissociation kinetics is to add oligonucleotides containing the specific site to a solution of a longer DNA fragment with prebound protein. Protein that dissociates from the DNA fragment is trapped by the added competitor. The ratio of specific site concentrations of oligonucleotide and of DNA fragment is at least 100 and is usually higher. At these high ratios, if protein was added to the mixture of the two, the probability that the protein will bind to the DNA fragment is less than 1%. We find, however, that the dissociation kinetics of both EcoRI and EcoRV depend on the oligonucleotide concentration. We surmise that this dependence is attributable to protein hopping kinetics. After the protein dissociates, it is still close to the DNA. The probability that the protein will rebind to the same DNA is quite large. Mathematical expressions are available for the distribution of hopping times. During this off-time the protein can bind to oligonucleotide. The probability that a protein will be captured by an oligonucleotide during a hopping excursion can be calculated from the oligonucleotide concentration, the association rate constant, and the hopping time distribution function. A distribution function determined by random walk simulations gives a reasonably good description of the oligonucleotide concentration dependence observed for EcoRI. The analytical expression for 2-dimensional first-passage times, however, predicts a much smaller dependence than is observed. The electrostatic attraction between protein and DNA may be the reason the analytic expression fails. Unlike EcoRI, the dependence of dissociation rate on oligonucleotide concentration for EcoRV is sensitive to pH. At low pH values, the oligonucleotide concentration dependence is about the same as for EcoRI while there is almost no dependence at pH 7.6. Our working hypothesis is that a return to the DNA does not necessarily mean a return to the recognition site; there is a probability that the protein will dissociate again before returning to the recognition site. In the limit of zero probability of rebinding to the specific sequence there will be no oligonucleotide concentration dependence. We have tested this hypothesis with EcoRI, increasing the dissociation rate of the nonspecifically bound protein by increasing salt concentration. We indeed observe that the overall dissociation rate of the complex loses its oligonucleotide concentration dependence. The pH sensitivity of EcoRV means either that the relative sliding and nonspecific dissociation rates are pH dependent or that the protein can dissociate in a pH–dependent conformation that is not able to rebind to DNA. This latter possibility is attractive given that the search process will be more efficient; the protein must diffuse further from the original binding region before being able to rebind to DNA.
Publications
- DeRouchey J, Parsegian VA, Rau DC. Cation charge dependence of the forces driving DNA assembly. Biophys J 2010;99:2608-2615.
- Qiu X, Parsegian VA, Rau DC. Divalent counterion condensation of triple-strand DNA. Proc Natl Acad Sci USA 2010;107:21482-21486.
- Qiu X, Rau DC, Parsegian VA, Fang LT, Knobler CM, Gelbart WM. Salt-dependent DNA-DNA spacings in intact bacteriophage lambda reflect relative importance of DNA self-repulsion and bending energies. Phys Rev Lett 2011;106:028102.
- Stanley C, Rau DC. Evidence for water structuring forces between surfaces. Curr Opin Colloid Interface Sci 2011;16:551-556.
- Sidorova NY, Muradymov S, Rau DC. Solution parameters modulating DNA binding specificity of the restriction endonuclease EcoRV. FEBS J 2011;278:2713-2727.
Collaborators
- Jason DeRouchey, PhD, University of Kentucky, Lexington, KY
- William Gelbart, PhD, University of California Los Angeles, Los Angeles, CA
- V. Adrian Parsegian, PhD, University of Massachusetts, Amherst, MA
- Xiangyun Qiu, PhD, George Washington University, Washington, DC
- T.J. Thomas, PhD, University of Medicine and Dentistry of New Jersey–Robert Wood Johnson Medical School, Piscataway, NJ
- William Whittier, DVM, Virginia-Maryland Regional College of Veterinary Medicine, Blacksburg, VA
Contact
For more information, email raud@mail.nih.gov.