

You are here: Home > Section on Molecular Transport
Biophysics of Large Membrane Channels

- Sergey M. Bezrukov, PhD, Head, Section on Molecular Transport
- Tatiana K. Rostovtseva, PhD, Staff Scientist
- Philip A. Gurnev, PhD, Research Fellow
- Oscar Teijido Hermida, PhD, Visiting Fellow
- Ekaterina M. Nestorovich, PhD, Contractor
- Kely L. Sheldon, MS, Predoctoral Intramural Research Training Award Fellow
We study mechanisms of transporter-facilitated metabolite exchange across cell and organelle membranes by combining physical theory and experiments on reconstitution of the corresponding "large" ion channels into planar lipid membranes. It is well-recognized that disturbances in metabolite transport result in a wide variety of pathologies in development and in different organ systems, including, among others, cancer, cystic fibrosis, seizure disorders, muscular dystrophies, and cardiomyopathies. Understanding of the physical mechanisms that govern metabolite transport is essential for creating new approaches to treatment of these pathologies.
To investigate channels under precisely controlled conditions, we first isolate the channel-forming proteins from their host organisms, then purify and reconstitute them into planar lipid membranes. Our main goal is to elucidate the physical principles and molecular mechanisms responsible for metabolite flux regulation under normal and pathological conditions. The channel-forming proteins of interest include the voltage-dependent anion channel from the outer membrane of mitochondria (VDAC), B-components of Clostridium botulinum C2 and Clostridium perfringens iota toxins, Bacillus anthracis protective antigen, Clostridium perfringens epsilon toxin, Escherichia coli general porin OmpF and sugar-specific LamB, Staphylococcus aureus alpha-hemolysin, Pseudomonas aeruginosa OprF, Trichoderma viride alamethicin, and Pseudomonas syringae lipopeptide toxin syringomycin E. We also use Bacillus brevis gramicidin A as a tool to study mechanical properties of lipid membranes.
Tailor-made beta-cyclodextrin derivatives as broad-spectrum inhibitors of binary pore-forming exotoxins
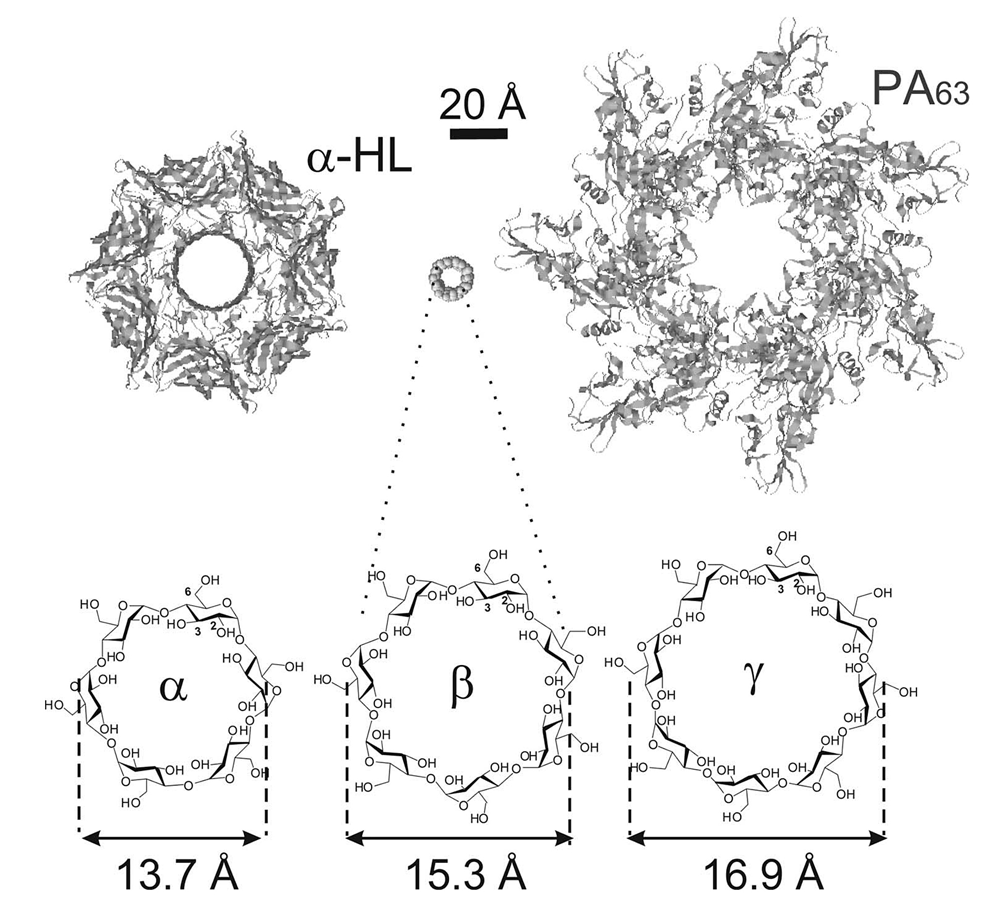
Click image to enlarge.
Figure 1.
Schematic illustration of the staphylococcal alpha-hemolysin channel (left) and anthrax PA (right) pre-pore in comparison with alpha-, beta-, and gamma-cyclodextrin molecules that were used as scaffolds to synthesize the effective blockers of "virulent" channels.
Clostridium botulinum C2 toxin and Clostridium perfringens iota toxin are binary exotoxins, which ADP-ribosylate actin in the cytosol of mammalian cells and thereby destroy the cytoskeleton. C2 and iota toxins consist of two individual proteins, an enzymatic active (A) component and a separate receptor binding and translocation (B) component. The latter forms a complex with the A component on the surface of target cells and, after receptor-mediated endocytosis, mediates the translocation of the A component from acidified endosomal vesicles into the cytosol. To this end, the B components form heptameric pores in endosomal membranes, which serve as translocation channels for the A components. We demonstrated that a seven-fold symmetrical positively charged beta-cyclodextrin derivative, per-6-S-(3-aminomethyl)benzylthio-beta-cyclodextrin, protects cultured cells from intoxication with C2 and iota toxins in a concentration-dependent manner starting at low micromolar concentrations. We established that the compound inhibited the pH–dependent membrane translocation of the A components of both toxins in intact cells. Consistently, the compound effectively blocked transmembrane channels formed by the B components of C2 and iota toxins in planar lipid bilayers in vitro in reconstitution experiments. With C2 toxin, we consecutively ruled out all other possible inhibitory mechanisms by showing that the compound did not interfere with the binding of the toxin to the cells or with the enzyme activity of the A component. In previous work, we identified the beta-cyclodextrin derivative as one of the most potent inhibitors of the binary lethal toxin of Bacillus anthracis both in vitro and in vivo, implying that the derivative might represent a broad-spectrum inhibitor of binary pore-forming exotoxins from pathogenic bacteria.
In a separate study, we compared the abilities of structurally related cationic cyclodextrins to inhibit Bacillus anthracis lethal toxin and Staphylococcus aureus alpha-hemolysin. We used alpha-, beta-, and gamma-cyclodextrins as scaffolds to synthesize cationic derivatives carrying six, seven, and eight positive charges, respectively. We found that, whereas alpha-cyclodextrins were ineffective, both beta- and gamma-cyclodextrin derivatives effectively inhibited anthrax toxin action by blocking the transmembrane oligomeric pores formed by the protective antigen (PA) subunit of the toxin. In contrast, the oligomeric pore of alpha-hemolysin was selectively blocked only by beta-cyclodextrin derivatives, demonstrating that, in addition to charge, both symmetry and size of the inhibitor and the pore are important.
Regulation of the voltage-dependent anion channel from the outer membrane of mitochondria by dimeric tubulin
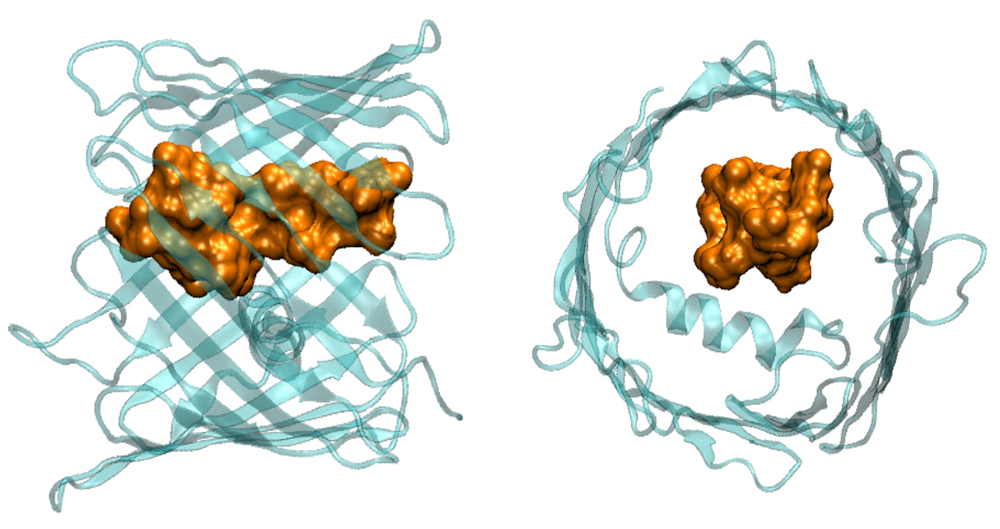
Click image to enlarge.
Figure 2.
Cartoon illustrating VDAC blockage by tubulin, in which the negatively charged tail (brown-yellow) of the tubulin heterodimer enters the predominantly positively charged VDAC pore (light blue).
In our previous work, we showed that one of the most abundant proteins in the cytosol of the majority of eukaryotic cells, dimeric tubulin, is a potent inhibitor of the VDAC from the outer mitochondrial membrane. For the channel, reconstituted into planar lipid membranes, the VDAC–tubulin interaction is seen as reversible transitions between the open and tubulin-blocked states. Experiments with isolated mitochondria and human hepatoma HepG2 cells demonstrated that VDAC–tubulin interaction is functionally important for regulating mitochondrial respiration. The tubulin-blocked state is still highly ion-conductive (about 40% of the open state conductance in 1 M KCl), which suggests that VDAC inhibition by tubulin is limited by the value of this residual conductance. It is important, however, that the major role of VDAC is regulation of ATP/ADP exchange between mitochondria and the cytosol rather than of the flux of small ions, so what really matters is the effect of tubulin blockage on the nucleotide transport. To assess the functional features of the tubulin-blocked state, we used three approaches. We first estimated the change in the characteristic radius of VDAC pore upon its blockage by tubulin using polymer partitioning, which is being developed in our laboratory. Based on the characteristic molecular weight of the polymer that separates partitioning from exclusion in VDAC's open and blocked states, we concluded that, as a result of the blockage, the effective cross-sectional area of the channel is reduced by a factor of two. Second, we analyzed the blockage-induced change in the channel small-ion selectivity at close to physiological salt concentrations. We showed that selectivity of the channel reverses its sign: from predominantly anionic selectivity in the open state, the channel shifts to cationic selectivity in the tubulin-blocked state. Third, we estimated ATP partitioning into both open and tubulin-blocked channel. We found that the addition of ATP reduces the conductance of the open-state channel but does not change conductance of the tubulin-blocked channel. We concluded that, as a result of electrostatic and steric barriers, ATP is excluded from the tubulin-blocked state of VDAC, thus establishing the functional role of the VDAC–tubulin interaction in regulating mitochondrial respiration.
Physical theory of facilitated metabolite transport: a functional role for transporter isoforms
Many of the nutrients a cell needs, such as sugars, amino acids, nucleotides or organic bases, require specialized transporters to cross the cell membrane. The rapid growth of available information, recently characterized as "transporter explosion", has led to creation of the transporter classification system, with division of all transporters into channels or carriers. This year, we focused on the physical principles of optimization of carrier-facilitated transport. The human genome harbors 43 distinct families of transport systems that comprise more than 300 isoforms of individual solute carriers. Although the majority of these transport systems are responsible for uptake of specific substrates, a substantial number of transporters are used for uptake of the same solute, and often there is overlapping expression of multiple isoforms in the same cell type. The question naturally arises as to why there are so many transporter isoforms. We found a possible answer to this question by analyzing carrier-facilitated transport with a focus on the optimal efficiency of the transporter. We demonstrated that, at lower substrate concentrations, stronger substrate binding is required and that the deviations from optimal interaction become more critical as the substrate concentration increases, i.e., higher concentrations necessitate more precise tuning. Thus, uniporters designed to transport the same molecule in the same cell have to be optimized with different amino-acid sequences, with one gene encoding a uniporter protein that functions most efficiently at high solute concentrations, while another gene encodes one that is most efficient at low concentrations. The existence of multiple transporter isoforms that carry the same molecule is well documented for almost any important substrate. Though this variety of isoforms may seem redundant and, in principle, could be explained by the lack of strong evolutionary pressures to decrease the size of the genome, our analysis now point to an alternative possibility. We have shown that transporter efficiency is fine-tuned to specific ranges of substrate concentration, so that different isoforms might be tailored to adjust their amino acid composition for the optimal strength of substrate/transporter interactions and the transition rates between various conformations.
Additional Funding
- NIAID intramural biodefense grant (2009-2011) "From anthrax to botulism, cholera, and epsilon toxin: in search for broad-spectrum antitoxins that block virulent nanopores"
Publications
- Berezhkovskii AM, Bezrukov SM. Effective drift and diffusion of a particle jumping between mobile and immobile states. J Electroanal Chem 2011;660:352-355.
- Berezhkovskii AM, Lizunov VA, Zimmerberg J, Bezrukov SM. Functional role for transporter isoforms in optimizing membrane transport. Biophys J 2011;101:L14-L16.
- Nestorovich EM, Karginov VA, Popoff MR, Bezrukov SM, Barth H. Tailor-made beta-cyclodextrin blocks the translocation pores of binary exotoxins from Clostridium botulinum and Clostridium perfringens and protects mammalian cells from intoxication. PLoS ONE 2011;6:e23927.
- Gurnev PA, Rostovtseva TK, Bezrukov SM. Tubulin-blocked state of VDAC studied by polymer and ATP partitioning. FEBS Lett 2011;585:2363-2366.
- Sheldon KL, Maldonado EN, Lemasters JJ, Rostovtseva TK, Bezrukov SM. Phosphorylation of voltage-dependent anion channel by serine/threonine kinases governs its interaction with tubulin. PLoS ONE 2011;6:e25539.
Collaborators
- Vicente M. Aguilella, PhD, Universidad Jaume I, Castellón, Spain
- Alexander M. Berezhkovskii, PhD, Division of Computational Bioscience, CIT, NIH, Bethesda, MD
- Susan K. Buchanan, PhD, Laboratory of Molecular Biology, NIDDK, Bethesda, MD
- Leonardo Dagdug, PhD, Universidad Autonoma Metropolitana-Iztapalapa, Mexico City, Mexico
- Vladimir A. Karginov, PhD, Innovative Biologics, Herndon, VA
- John J. Lemasters, PhD, Medical University of South Carolina, Charleston, SC
- Adrian Parsegian, PhD, Program in Physical Biology, NICHD, Bethesda, MD
- Dan Sackett, PhD, Program in Physical Biology, NICHD, Bethesda, MD
- Gerhard Wagner, PhD, Harvard Medical School, Cambridge, MA
- Michael Weinrich, MD, National Center for Medical Rehabilitation Research, NICHD, Bethesda, MD
- Josh Zimmerberg, MD, PhD, Program in Physical Biology, NICHD, Bethesda, MD
Contact
For more information, email bezrukov@helix.nih.gov or visit smt.nichd.nih.gov.