

You are here: Home > Section on Organelle Biology
Dynamics of Membrane Trafficking, Sorting, and Compartmentalization Within Eukaryotic Cells

- Jennifer Lippincott-Schwartz, PhD, Head, Section on Organelle Biology
- Dylan Burnette, PhD, Postdoctoral Fellow
- Brian Daniels, PhD, Postdoctoral Fellow
- Carolyn Ott, PhD, Postdoctoral Fellow
- Angelika Rambold, PhD, Postdoctoral Fellow
- Prasanna Satpute, PhD, Postdoctoral Fellow
- Prabuddha Sengupta, PhD, Postdoctoral Fellow
- Arnold Seo, PhD, Postdoctoral Fellow
- Tijana Talisman, PhD, Postdoctoral Fellow
- Schuyler Van Engelenburg, PhD, Postdoctoral Fellow
- Ernesto Ambroggio, PhD, Visiting Fellow
- Yu Chen, PhD, Visiting Fellow
- Sarah Cohen, PhD, Visiting Fellow
- Natalie Elia, PhD, Visiting Fellow
We investigate the global principles underlying secretory membrane trafficking, sorting, and compartmentalization within eukaryotic cells. We use live-cell imaging of green fluorescent protein (GFP) fusion proteins in combination with photobleaching and photoactivation techniques to investigate the subcellular localization, mobility, transport routes, and turnover of a variety of proteins with important roles in the organization and regulation of membrane trafficking and compartmentalization. To test mechanistic hypotheses about protein and organelle dynamics, we use quantitative measurements of these protein characteristics in kinetic modeling and simulation experiments. Among the topics under investigation are (i) membrane partitioning and its role in protein sorting and transport in the Golgi apparatus; (ii) biogenesis and dynamics of unconventional organelles; (iii) mitochondrial morphology and its regulation of cell cycle progression; and (iv) cytoskeletal and endomembrane cross-talk in polarized epithelial cells, hematopoietic niche cells, and the developing Drosophila syncytial blastoderm embryo. We also devoted considerable effort to developing new fluorescence microscopy techniques for imaging fluorescently tagged proteins at near-molecular resolution.
Development of green fluorescent protein technology

Jennifer Lippincott-Schwartz lab photo
(click image to enlarge)
Super-resolution techniques such as photoactivated localization microscopy (PALM) permit the imaging of fluorescent protein chimeras, revealing the organization of genetically expressed proteins on the nanoscale with a density of molecules high enough to provide structural context. The PALM method involves serial photoactivation and subsequent bleaching of numerous sparse subsets of photoactivated fluorescent protein molecules. A statistical fit of the point-spread function of individual molecules' centers of fluorescent emission then localizes the molecules at near-molecular resolution. The aggregate position information from all subsets is then assembled into a super-resolution image that isolates individual fluorescent molecules at high molecular densities (up to 105 molecules/µm2). We previously demonstrated PALM imaging of intracellular structures (including lysosomes, Golgi apparatus, and mitochondria) in cryo-prepared thin sections as well as imaging of vinculin and actin in fixed cells with TIRF (total internal reflection fluorescence) excitation along with correlative PALM/transmission electron microscopy of a mitochondrial marker protein. We recently helped develop a new photoactivatable fluorescent protein called PA-mCherry that switches from dark to red upon UV illumination. Using PA-mCherry and PA-GFP, we demonstrated two-color photoactivation imaging. The two-color approach was also used for PALM imaging of two different proteins within cells. We also used PALM in a new technique for single-particle tracking (sptPALM) in living cells, permitting characterization of protein diffusion and immobilization at the single-molecule level. We further developed a pair-correlation PALM (PC-PALM) approach that enables one to determine whether proteins on the plasma membrane are randomly distributed or exist in clusters. If clustered, the approach permits determination of the cluster size and protein density within the cluster. The PC-PALM technique can provide novel insights into how proteins organize themselves for signaling and other activities at the plasma membrane. We also developed a method for counting single molecules in PALM images in order to decipher subunit stoichiometry within receptor complexes.
Membrane partitioning and its role in protein sorting and transport within the Golgi apparatus
The Golgi apparatus processes and filters newly synthesized protein and lipid moving through the secretory pathway, but how it retains resident components in the face of forward flux of cargo is not completely understood. To gain insight into this question, we used a rapamycin-based trapping protocol for capture of Golgi enzymes in the endoplasmic reticulum (ER) to address the extent of Golgi enzyme cycling through the ER. In cells expressing an FRB (red fluorescent protein)–tagged Golgi enzyme marker and FKBP (FK506-binding protein)–tagged ER protein, the Golgi marker completely redistributed into the ER within 2 hours of rapamycin treatment. An FRB-tagged photoactivatable Golgi enzyme, highlighted uniquely in the Golgi, likewise redistributed to the ER upon rapamycin treatment, revealing that Golgi protein signal in the ER originates from a Golgi pool. Rapamycin-trapped fluorescent FRAP (fluorescence recovery after photobleaching)–Golgi markers and fluorescent FKBP-ER markers underwent fluorescence resonance energy transfer (FRET) in the ER, demonstrating direct binding between the two markers upon redistribution. Inefficient rapamycin-induced ER trapping occurred with Golgi enzymes tagged with FKBP in cells expressing FRB–tagged ER proteins. This was attributable to an endogenous ER-localized FKBP, which competes with the FKBP-Golgi enzyme for binding to the FRB–tagged ER protein, explaining previous negative results. Using FRB–tagged Golgi enzyme and FKBP–tagged ER protein under rapamycin treatment to characterize Golgi enzyme recycling to the ER, we showed that the pathway is blocked by PLA2 inhibitors and by expression of a dominant negative Rab6 mutant. Moreover, Golgi fragmentation during microtubule depolymerization is dependent on Golgi enzyme cycling through the ER. Thus, Golgi structure and maintenance are fundamentally reliant on the ER.
Biogenesis and dynamics of unconventional organelles
We explored the dynamics of several organelles, including autophagosomes, primary cilia, and midbodies. Autophagosomes form during autophagy, which is a highly conserved, bulk degradation pathway that is also involved in turnover of large aggregates and organelles within cells. While emerging results have revealed the importance of autophagy in various biological and pathological processes, how this pathway operates is far from clear. We used various live cell–imaging and molecular-genetic approaches to investigate the membrane origin of autophagosomes and the signals that recruit substrates to this organelle. Our data revealed that the outer membrane of mitochondria serves as the membrane source during starvation-induced autophagy formation and maturation. Furthermore, ubiquitin modification acts as a targeting signal for delivery of small cytosolic proteins and larger organelles to autophagosomes. We further discovered that mitochondria extensively tubulate under starvation conditions due to a shift in the fission/fusion dynamics of these organelles toward fusion. This was dependent on the dynamin-related protein (DRP1) becoming phosphorylated under nutrient deprivation. The highly tubulated mitochondria were more resistant to mitophagy, the process whereby mitochondria are destroyed through autophagy.
The primary cilium is a chemosensory and mechanosensory organelle that receives signals (both chemical and mechanical) from the extracellular environment. We imaged live polarized epithelial cells and found that cilia of one cell can make direct contact (bridges), which can be stable over many hours, with cilia of adjacent or nearby cells. The bridges are composed of cilia from individual cells; that is, they are not a continuation from one cell to the next. Cilia can thus initiate and mediate cell-cell communication through direct contact.
The midbody is the central region of the narrow membrane bridge that connects two daughter cells during cytokinesis. Cleavage of the narrow membrane neck on either side of the midbody is the final step in cytokinesis. We used structured illumination and time-lapse microscopy to clarify midbody cleavage, focusing on the role of endosomal sorting complexes required for transport (ESCRT). We found that the upstream ESCRT-I subunit TSG101 localizes to two cortical rings adjacent to the midbody center. The ESCRT-III subunit CHMP4B recruits to these rings, but at late stages in abscission also co-localizes with the narrow constriction at the site of membrane cutting. Direct visualization of the kinetics of ESCRT recruitment revealed an ordered assembly mechanism. The midbody protein CEP55 arrives early in cytokinesis, followed by ESCRT-I (TSG101), ESCRT-III (CHMP4B), and the ESCRT disassembly factor VPS4. The arrival of ESCRT-III and VPS4 coincides in time and space with cell separation, consistent with a direct role for these proteins in membrane abscission.
Mitochondrial morphology and its regulation of cell cycle progression

Click image to enlarge.
Figure 1. Mitochondria morphology in NRK cells
Image, obtained by structured illumination microscopy, of mitochondria labeled with GFP-tagged prohibitin.
Mitochondria are important energy-producing organelles within cells. Their morphology, including fragmented elements and tubular networks, results from a balance between fission and fusion events. To determine whether changes occur in mitochondrial dynamics at various stages of the cell cycle, we carried out live-cell imaging experiments in cells stably expressing RFP (red fluorescent protein) targeted to the mitochondrial matrix. We found that mitochondria exhibit distinct morphological and physiological states at different stages of the cell cycle. During mitosis, mitochondria fragment into hundreds of small units for partitioning into daughter cells at cytokinesis. Strikingly, at G1/S, mitochondria fuse into a single, huge, dynamic filamentous system, unlike at any other stage of the cell cycle. Photobleaching of an area across the filamentous system revealed that the mitochondrial matrix is continuous. In addition, the mitochondrial network is electrically coupled and has a higher membrane potential than mitochondria at any other stage of the cell cycle. When the filamentous network or its membrane potential was disrupted or its dynamics perturbed, cell cycle progression from G1 into S phase was arrested in a p53-dependent manner. Moreover, p21 overexpression, which induces a G1/S arrest, resulted in filamentous mitochondria with reduced matrix continuity and loss of electrical coupling. The data revealed that, during the cell cycle, mitochondrial dynamism and morphology undergo critical changes that are sensed by the cell at G1/S to control cell cycle progression.
Cytoskeletal dynamics at the cell's leading edge and its role in cell crawling
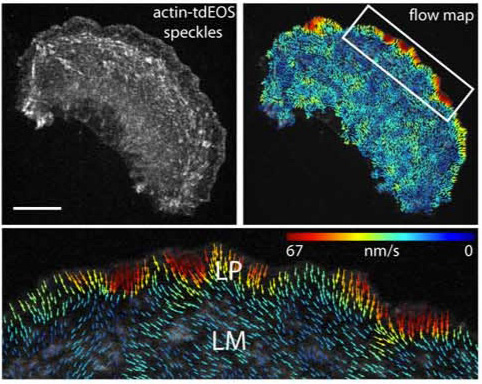
Click image to enlarge.
Figure 2. Retrograde actin flow rates
Actin-tdEOS speckle image of an entire PtK1 cell with a corresponding flow map and a higher magnification of the leading edge flow. Vector colors reflect flow speed (color bar), and arrows reflect direction. Scale bar (upper left panel) 10 μm.
Epithelial cell migration requires coordination of two actin modules at the leading edge: one in the lamellipodium and one in the lamella. How the two modules connect mechanistically to regulate directed edge motion is not understood. Using live-cell imaging and photoactivation approaches, we demonstrated that the actin network of the lamellipodium evolves spatio-temporally into the lamella. This occurs during the retraction phase of edge motion, when myosin II redistributes to the lamellipodial actin and condenses it into an actin arc parallel to the edge. The new actin arc moves rearward, slowing down at focal adhesions in the lamella. Based on these observations, we proposed that net edge extension occurs by nascent focal adhesions advancing the site at which new actin arcs slow down and form the base of the next protrusion event. In this model, the actin arc thereby serves as a structural element underlying the temporal and spatial connection between the lamellipodium and the lamella during directed cell motion.
Additional Funding
- IATAP (Intramural AIDS Targeted Antiviral Program)
- Director Challenge Award
Publications
- Sengupta P, Jovanovic-Talisman T, Skoko D, Renz M, Veatch SL, Lippincott-Schwartz J. Probing protein heterogeneity in the plasma membrane using PALM and pair correlation analysis. Nat Methods 2011;8:969-975.
- Rambold AS, Kostelecky B, Elia N, Lippincott-Schwartz J. Tubular network formation protects mitochondria from autophagosomal degradation during nutrient starvation. Proc Natl Acad Sci USA 2011;108:10190-10195.
- Burnette DT, Manley S, Sengupta P, Sougrat R, Davidson MW, Kachar B, Lippincott-Schwartz J. A role for actin arcs in the leading-edge advance of migrating cells. Nat Cell Biol 2011;13:371-381.
- Elia N, Sougrat R, Spurlin TA, Hurley JH, Lippincott-Schwartz J. Dynamics of endosomal sorting complex required for transport (ESCRT) machinery during cytokinesis and its role in abscission. Proc Natl Acad Sci USA 2011;108:4846-4851.
- Fu D, Wakabayashi Y, Lippincott-Schwartz J, Arias IM. Bile acid stimulates hepatocyte polarization through a cAMP-Epac-MEK-LKB1-AMPK pathway. Proc Natl Acad Sci USA 2011;108:1403-1408.
Collaborators
- Win Arias, MD, Cell Biology and Metabolism Program, NICHD, Bethesda, MD
- Michael Davidson, PhD, Florida State University, Tallahassee, FL
- Harald Hess, PhD, Howard Hughes Medical Institute, Janelia Farm Campus, Ashburn, VA
- Robert Phair, PhD, BioInformatics, Rockville, MD
- Vladimir Verkhusha, PhD, Albert Einstein School of Medicine, Bronx, New York
Contact
For more information, email lippincj@mail.nih.gov or visit lippincottschwartzlab.nichd.nih.gov.