

You are here: Home > Section on Cellular Differentiation
Molecular Genetics of Heritable Human Disorders

- Janice Y. Chou, PhD, Head, Section on Cellular Differentiation
- Hyun-Sik Jun, PhD, Staff Scientist
- Yong Jun Lee, PhD, Visiting Fellow
- Young Mok Lee, PhD, Visiting Fellow
- Su Ru Lin, PhD, Visiting Fellow
- Brian C. Mansfield, PhD, Guest Researcher
- Chi-Jiunn Pann, BS, Senior Research Assistant
- Lane H. Wilson, BS, Postbaccalaureate Fellow
We conduct research to delineate the pathophysiology of glycogen storage disease type I (GSD-I) and glucose-6-phosphatase-beta (G6Pase-beta or G6PC3) deficiency and to develop novel therapies for these disorders. GSD-I consists of two subtypes, GSD-Ia, deficient in G6Pase-alpha (or G6PC), and GSD-Ib, deficient in the glucose-6-phosphate transporter (G6PT). A third disease, G6Pase-beta deficiency, also known as severe congenital neutropenia syndrome type 4 (SCN4), is also grouped with GSD-I. G6Pase-alpha and G6Pase-beta are endoplasmic reticulum (ER)–bound glucose-6-phosphate (G6P) hydrolases. Their active sites lie inside the lumen, which depend upon G6PT to translocate G6P from the cytoplasm into the ER lumen. The G6PT/G6Pase-alpha complex maintains interprandial glucose homeostasis. The G6PT/G6Pase-beta complex maintains energy homeostasis and the functionality of neutrophil and macrophages. GSD-Ia and GSD-Ib patients manifest a common metabolic phenotype of impaired glucose homeostasis not shared by G6Pase-beta deficiency while GSD-Ib and G6Pase-beta–deficient patients manifest a common myeloid phenotype of neutropenia and myeloid dysfunction not shared by GSD-Ia. Neutrophils express the G6PT/G6Pase-beta complex, and inactivation of G6PT or G6Pase-beta leads to enhanced neutrophil apoptosis, which underlies neutropenia in GSD-Ib and G6Pase-beta deficiency. Recently, we showed that G6Pase-beta is essential for energy homeostasis in neutrophils and macrophages. G6Pase-beta deficiency prevents recycling of ER glucose to the cytoplasm, leading to neutrophil/macrophage dysfunction. There is no cure for either GSD-I or G6Pase-beta deficiency. Animal models of the three disorders are available and are being exploited to both delineate the disease more precisely and develop new treatment approaches, including gene therapy.
G-CSF improves G6Pase-beta–deficient neutrophil function by modulating apoptosis and energy homeostasis.
G6Pase-beta deficiency underlies a congenital neutropenia syndrome in which neutrophils exhibit elevated ER stress, increased apoptosis, and impaired energy homeostasis and functionality. Granulocyte colony–stimulating factor (G-CSF), a cytokine that is widely used to treat neutropenia, can delay neutrophil apoptosis by modulating apoptotic mediators. In G6Pase-beta deficiency, G-CSF is used to improve neutrophil counts and thus reduce the number and severity of bacterial infections but its impact on neutrophil apoptosis and dysfunction is unknown. We hypothesized that in neutrophils of G6Pase-beta–deficient (G6pc3-/-) mice, G-CSF would both delay apoptosis and stimulate glucose uptake, which in turn would improve neutrophil energy homeostasis and functionality. Our results support this hypothesis. We showed that, in vitro, G-CSF modulates apoptotic mediators and delays apoptosis in G6pc3-/- neutrophils, although G6pc3-/- neutrophils still exhibit accelerated apoptosis compared with wild-type neutrophils. In contrast, in vivo G-CSF therapy completely corrects neutropenia and normalizes levels of active caspase-3. Moreover, neutrophils from in vivo G-CSF–treated G6pc3-/- mice exhibit increased glucose uptake and elevated intracellular levels of G6P, lactate, and ATP, leading to improved functionality. Together, our results show that G-CSF improves G6pc3-/- neutrophil survival by modulating apoptotic mediators and rectifies function by enhancing energy homeostasis, providing insights into the etiology of neutropenia and neutrophil dysfunction in G6Pase-beta deficiency. We also delineated the signaling pathways of ER stress and apoptosis in G6pc3-/- neutrophils. We found evidence that protein kinase–like ER kinase–mediated signaling is one pathway that mediates ER stress and that the intrinsic mitochondrial pathway mediates, in part, neutrophil apoptosis in G6pc3-/- neutrophils.
AAV–mediated gene therapy prevents HCA and corrects metabolic abnormalities in murine GSD-Ia.
GSD-Ia patients deficient in G6Pase-alpha (or G6PC) manifest impaired glucose homeostasis. There is no cure for GSD-Ia, but many of the disease symptoms can be managed or improved using dietary therapies to maintain normoglycemia. While the therapies are sufficiently successful to enable patients to attain near normal growth and pubertal development, the underlying pathological process remains uncorrected. As a result, long-term complications including hepatocellular adenoma (HCA) with malignant potential still persist in GSD-Ia patients. Using our G6pc-/- mouse model of GSD-Ia, we examined the efficacy of liver G6Pase-alpha delivery mediated by AAV8-G6PC-GPE, an AAV serotype 8 vector expressing human G6Pase-alpha directed by the human G6PC promoter/enhancer (GPE) and showed that AAV8-G6PC-GPE–mediated gene transfer completely normalizes hepatic G6Pase-alpha deficiency for at least 24 weeks. A remaining concern with the therapy is the risk of HCA. A recent study showed that HCA develops in 100% of liver-specific G6pc-null mice 78 weeks after gene deletion. Therefore we examined the disease risk for hepatic neoplasia in a long-term study using G6pc-/- mice. We showed that AAV8-G6PC-GPE-mediated gene therapy maintains efficacy for at least 70-90 weeks for mice expressing more than 3% of wild-type hepatic G6Pase-alpha activity. The treated mice displayed normal hepatic fat storage, normal blood metabolite and glucose tolerance profiles, and reduced fasting blood insulin levels and maintained normoglycemia over a 24-hour fast. Ultrasound and histological examinations showed no hepatic steatosis or detectable HCA or hepatocellular carcinoma (HCC) in the liver of any of these mice. After a 24-hour fast, hepatic G6PT mRNA levels in AAV8-GPE-treated G6pc-/- mice were markedly elevated. The fact that G6PT transport is the rate-limiting step in microsomal G6P metabolism may explain why the treated G6pc-/- mice could sustain prolonged fasts. The low fasting blood insulin levels and lack of hepatic steatosis may explain the absence of HCA. Our results suggest that G6Pase-alpha gene transfer offers a therapeutic approach to the management of human GSD-Ia.
G6Pase-beta is essential for energy homeostasis in macrophages and the pregnant uterus, a deficiency leading to reduced fertility in G6pc3-/- mothers.
In addition to neutropenia and neutrophil dysfunction, G6Pase-beta–deficient patients also exhibit a distinct phenotype of increased visibility of superficial veins, congenital heart defects, and urogenital malformations, suggesting that G6Pase-beta deficiency underlies a broader cell dysfunction. The ubiquitous pattern of G6Pase-beta expression suggests that G6Pase-beta plays a critical role in non-gluconeogenic tissues where there may be increased demands for glucose. Macrophages play key roles in innate immunity, inflammation, and tissue remodeling. During pregnancy, macrophages also influence the homeostasis of the developing placenta and are important in preventing premature fetal rejection. Like neutrophils, macrophages have an elevated demand for glucose, and pregnancy is a condition with an elevated demand for glucose. We hypothesized that the cycling pathway for G6P metabolism (Figure 1) also occurs in macrophages and the pregnant uterus and thus that G6Pase-beta deficiency is associated with impaired energy homeostasis in both macrophages and the pregnant uterus, resulting in macrophage dysfunction and pregnancy-associated complications; this proved to be the case. We showed that murine G6pc3-/- macrophages exhibit impairments in their respiratory burst, chemotaxis, calcium flux, and phagocytic activities. Consistent with a G6P metabolism deficiency, G6pc3-/- macrophages also have lower glucose uptake and lower levels of G6P, lactate, and ATP than wild-type macrophages; furthermore, the expression and activation of NADPH oxidase activity is down-regulated. We further showed that, during pregnancy, the absence of G6Pase-beta activity also impaired energy homeostasis in the uterus, resulting in reduced production of monocyte chemoattractant protein 1 and macrophage colony-stimulating factor, cytokines that recruit macrophages to a site of injury or inflammation. Supporting this notion, G6pc3-/- macrophages exhibit repressed trafficking in vivo both during an inflammatory response and in pregnancy. The dysfunctional macrophages and impaired uterine energy homeostasis correlate with reduced fertility in G6pc3-/- mothers.
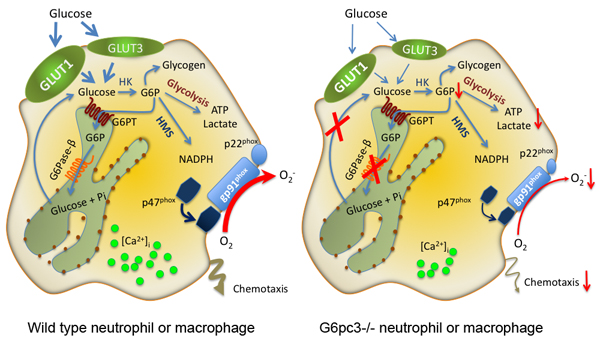
Figure 1. A schematic representation elucidating the cycling pathway for G6P metabolism in neutrophils/macrophages
G6P participates in four major pathways: glycolysis, hexose monophosphate shunt (HMS), glycogen synthesis, or ER cycling. In cycling, G6P enters the ER via G6PT, where it can accumulate until it is hydrolyzed to glucose by G6Pase-beta and transported back into the cytoplasm. By limiting the cytoplasmic glucose/G6P availability, cycling regulates the other three cytoplasmic pathways for G6P metabolism. Disruption of this cycling in G6Pase-beta–deficient neutrophils/macrophages results in impaired energy homeostasis and functionality.
SLC37A1 and SLC37A2 are phosphate-linked G6P antiporters.
Blood glucose homeostasis between meals depends upon production of glucose within the ER of the liver and kidney by hydrolysis of G6P into glucose and phosphate (Pi). The reaction depends on coupling the G6PT with G6Pase-alpha. G6PT belongs to the SLC37 family of membrane-bound transporters. Only G6PT, also known as SLC37A4, has been characterized; it acts as a Pi-linked G6P antiporter. The other three SLC37 family members, predicted to be sugar-phosphate:Pi exchangers, have not been characterized functionally. We therefore investigated whether any of these proteins might also have a G6P transport activity. Using reconstituted proteoliposomes, we examined the antiporter activity of the other SLC37 members along with their ability to couple with G6Pase-alpha. We showed that SLC37A1 and SLC37A2 are ER-associated, Pi-linked antiporters that can transport G6P. The activity of SLC37A3 is unknown. Unlike G6PT, neither is sensitive to chlorogenic acid, a competitive inhibitor of physiological ER G6P transport, and neither couples to G6Pase-alpha. We conclude that three of the four SLC37 family members are functional sugar-phosphate antiporters. However, only G6PT/SLC37A4 matches the characteristics of the physiological ER G6P transporter, suggesting the other SLC37 proteins do not play roles in blood glucose homeostasis.
Additional Funding
- The Childrens' Fund for Glycogen Storage Disease Research, 2011, ongoing.
Publications
- Jun HS, Lee YM, Song KD, Mansfield BC, Chou JY. G-CSF improves murine G6PC3-deficient neutrophil function by modulating apoptosis and energy homeostasis. Blood 2011;117:3881-3892.
- Pan CJ, Chen SY, Jun HS, Lin SR, Mansfield BC, Chou JY. SLC37A1 and SLC37A2 are phosphate-linked, glucose-6-phosphate antiporters. PloS ONE 2011;6(9):e231.
- Jun HS, Cheung YY, Lee YM, Mansfield BC, Chou JY. Glucose-6-phosphatase-β, implicated in a congenital neutropenia syndrome, is essential for macrophage energy homeostasis and functionality. Blood 2012;119:4047-4055.
- Lee YM, Jun HS, Pan CJ, Lin SR, Wilson LH, Mansfield BC, Chou JY. Prevention of hepatocellular adenoma and correction of metabolic abnormalities in murine glycogen storage disease type Ia by gene therapy. Hepatology 2012; in press.
- Chou JY, Jun HS, Mansfield BC. The SLC37 family of phosphate-linked sugar phosphate antiporters. Mol Aspects Med 2012; in press.
Collaborators
- Alessandra Eva, PhD, Gaslini Institute, Genova, Italy
- Philip M. Murphy, MD, Laboratory of Host Defenses, NIAID, Bethesda, MD
- Luigi Varesio, PhD, Gaslini Institute, Genova, Italy
- David A. Weinstein, MD, University of Florida College of Medicine, Gainesville, FL
Contact
For further information, contact chouja@mail.nih.gov.