

You are here: Home > Section on Growth and Development
Regulation of Childhood Growth

- Jeffrey Baron, MD, Head, Section on Growth and Development
- Kevin Barnes, PhD, Senior Research Assistant
- Julian Lui, PhD, Postdoctoral Fellow
- Youn Hee Jee, MD, Clinical Fellow
- Fariha Kamran, MD, Clinical Fellow
- Sao Fong Cheung, Predoctoral Fellow
- Stephan S. Spath, PhD, Special Volunteer
We investigate the cellular and molecular mechanisms governing childhood growth and development. We are especially interested in mechanisms that allow rapid cell proliferation and hence rapid body growth in young mammals and that subsequently suppress proliferation, thus setting a fundamental limit on the adult body size of the species. One goal of this work is to gain insight into the many human genetic disorders that cause childhood growth failure or overgrowth. In addition, further investigation of the identified growth-limiting mechanisms may lead to broader medical applications, because disruption of these mechanisms may contribute to oncogenesis, and conversely transient therapeutic suspension of growth-limiting mechanisms in adult cells might be used to achieve tissue regeneration.
Identification of genes that regulate the human growth plate
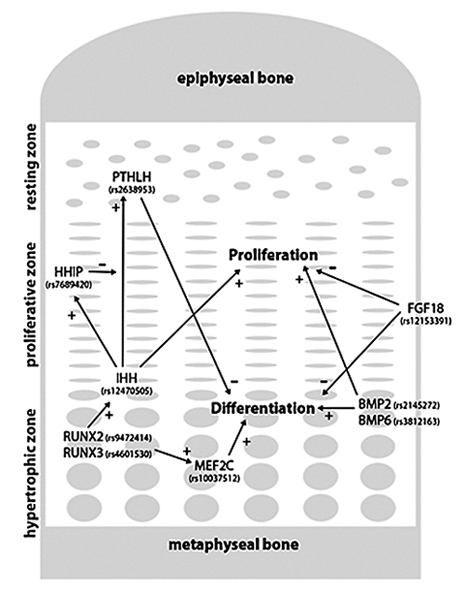
Figure 1. Examples of genes from GWAS analysis that have a well established role in chondrogenesis
PTHrP (encoded by PTHLH) produced in the resting zone inhibits differentiation of proliferative chondrocytes. The production of PTHrP is, in turn, stimulated by IHH from the hypertrophic zone. IHH stimulates chondrocyte proliferation in proliferative zone and the expression of HHIP, which inhibits and modulates the action of IHH in the resting zone. Together, IHH and PTHrP form a negative feedback loop that determines the height of the proliferative zone. BMP2 and BMP6, produced by hypertrophic chondrocytes, stimulate proliferation and differentiation of chondrocytes, processes that are partially antagonized by an inhibitory effect of FGFs, such as FGF18. RUNX2 and RUNX3 are expressed in the hypertrophic zone and stimulate hypertrophic differentiation through one of their downstream targets, MEF2C, which is also highly expressed in the hypertrophic zone. RUNX2 also interacts with the IHH–PTHrP feedback loop by inducing IHH expression. +, stimulatory effect; –, inhibitory effect
The growth plate is a thin layer of cartilage that is responsible for longitudinal bone growth and therefore determines height. It is composed of three principal zones: the resting zone, the proliferative zone, and the hypertrophic zone. We previously provided evidence that resting-zone chondrocytes serve as progenitor cells that can generate new clones of proliferative-zone chondrocytes. The proliferative chondrocytes replicate rapidly but then terminally differentiate to form the hypertrophic zone, which is invaded by blood vessels, osteoclasts, and differentiating osteoblasts that remodel the newly formed cartilage into bone tissue. The combined process of chondrogenesis and ossification leads to progressive lengthening of the bone. The growth plate undergoes important temporal development—embryonic formation, followed by a gradual, postnatal, programmed involution and loss of function, which causes the rate of bone growth to decrease and approach zero as the organism approaches its adult size, a process known as growth plate senescence.
Chondrogenesis in the growth plate requires several interacting regulatory systems. Our group has combined expression analysis and functional studies to help elucidate growth plate regulation by fibroblast growth factors (FGFs), bone morphogenetic proteins (BMPs), C-type natriuretic peptide, retinoids, WNTs, parathyroid hormone–related peptide (PTHrP)/Indian hedgehog (IHH), insulin-like growth factors (IGFs), estrogens, and glucocorticoids. However, such studies by our group and those of others generally use rodent models. Consequently, much less is known about the regulation of the human growth plate.
One powerful approach to investigate human linear growth involves genome-wide association studies (GWAS). A recent GWAS meta-analysis identified 180 loci that influence adult height. Each locus implies the presence of a nearby gene that plays a biological role in human linear growth. However, at each locus there are several genes that are sufficiently close to account for the linkage to height, and it is unclear which of these genes modulate height and which are innocent neighbors. We developed a new analytic method to partially address this deficiency.
We reasoned that many of the causative genes within the GWAS loci influence height because they are expressed in and function in the growth plate, the structure that causes elongation of long bones and vertebral bodies and thus determines height. Therefore, to identify genes that contribute to human height variation by acting in the growth plate, we used human disease databases, a mouse knockout phenotype database, and expression microarray studies of mouse and rat growth plate to identify genes that are likely required for normal growth plate function. We then searched for these growth plate–related genes within the height-determining loci defined by GWAS. Individually, each method identified significantly more genes within the GWAS height loci than at random genomic locations, supporting the validity of the approach. Together, these functional and expression analyses implicated 78 genes from the GWAS loci.
Many of the 78 implicated genes participate in molecular pathways that are known to be important for growth plate chondrogenesis in the mouse. Many participate in signaling pathways that regulate growth plate chondrocyte proliferation and differentiation, including those that encode IHH, PTHrP, the BMP/transforming growth factor-beta (TGFbeta) superfamily, C-type natriuretic peptide, FGF, and IGF signaling (Figure 1). Other implicated genes are involved in the formation of growth plate cartilage extracellular matrix, including structural proteins in the cartilage matrix (ACAN, COL11A1), as well as genes that encode enzymes involved in matrix biosynthesis (GFPT2, SERPINH1) and degradation (GALNS, ADAMTS10). The analysis also identified several genes involved in cell-cycle processes, including DNA synthesis (TOP2A), mitotic spindle formation (CENPW, TACC3), cell-cycle progression (E2F1), and translation of replication-dependent histones (SLBP). Altering the function of such genes might affect cell proliferation not just in growth plate but also in other tissues. As might be expected, the genes were typically found to be expressed at high levels in the proliferative zone and to be subsequently downregulated in the hypertrophic zone, in which chondrocyte replication ceases.
One of the most exciting outcomes of this analysis is that it implicates many genes not previously known to regulate either the mouse or human growth plate. For example, IGF2BP2 was implicated because it shows higher expression in growth plate cartilage than in other tissues and because its expression is downregulated during growth plate senescence. Similarly, IGF2BP3 was implicated because its expression is upregulated during hypertrophic differentiation and downregulated during growth plate senescence. Neither gene has a known human or mouse phenotype, but targeted ablation of IGF2BP1, the third member of the gene family, causes shortened bones with advanced mineralization. Thus, taken together, the data suggest that this family of proteins plays an important regulatory role in both the rodent and human growth plate, perhaps through their ability to bind to specific mRNAs, including IGF2.
Another important implication of the current analysis is that it identifies strong candidates for the etiology of human growth disorders, including isolated short stature, skeletal dysplasias, and short stature that is part of a larger dysmorphic syndrome. Our analysis suggests that, for many of the height-associated genes, common polymorphisms subtly affect gene expression or function in the growth plate and thus influence adult height. Uncommon mutations in these genes that have a greater effect on expression or function would therefore be expected to cause greater disturbances in growth plate chondrogenesis, resulting in short stature or a skeletal dysplasia. Indeed, the height-associated loci were highly enriched with genes that cause skeletal dysplasias when mutated in humans. Thus, we think it likely that other growth plate–related genes identified by this analysis also cause short stature and skeletal dysplasias of unknown etiology.
Imprinted genes that affect body size contribute to growth of embryonal malignancies.
Click image to enlarge.
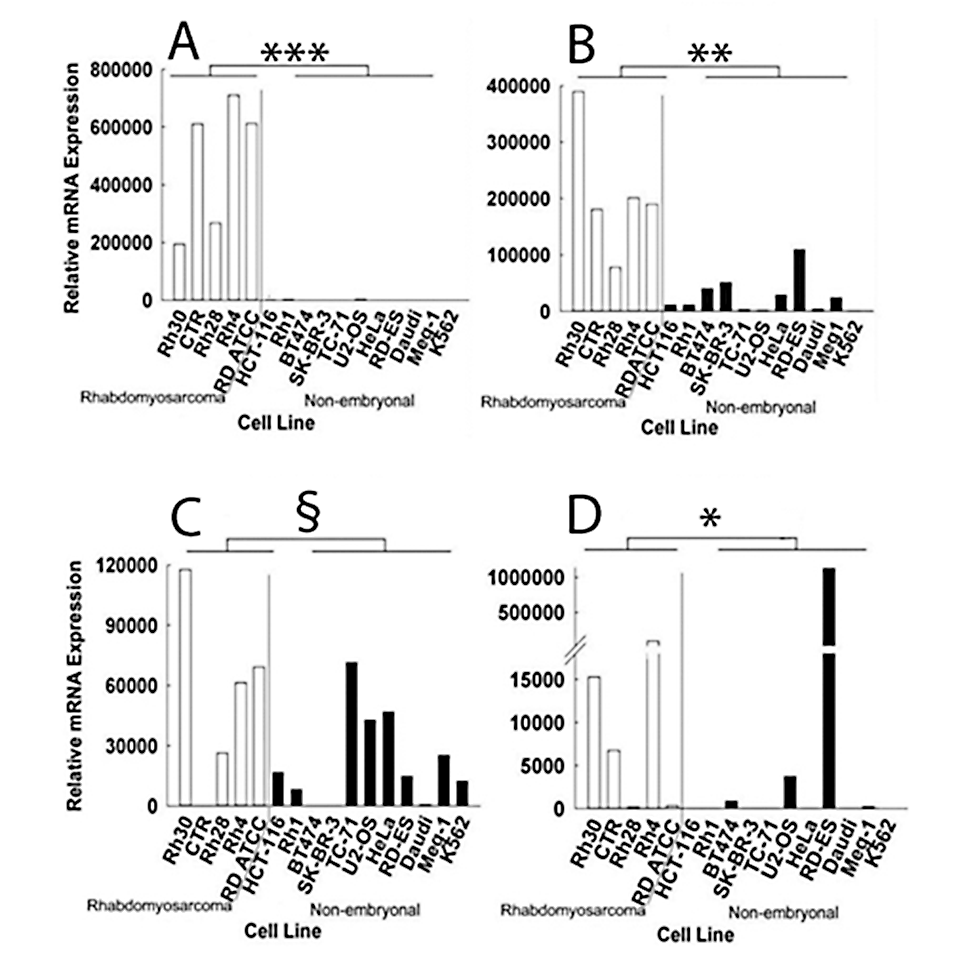
Figure 2. Relative mRNA expression of IGF2 (A), MEST (B), PLAGL1 (C), and PEG3 (D) in rhabdomyosarcoma cell lines and non-embryonal cancer cell lines, measured by real-time PCR
Levels of IGF2, MEST, and PEG3 mRNA were significantly higher in the 5 rhabdomyosarcoma cell lines than in 11 non-embryonal cancer cell lines. PLAGL1 did not reach significance. White bars, relative mRNA expression in rhabdomyosarcoma cell lines; black bars, relative mRNA expression in non-embryonal cancer cell lines. *, P = 0.05; **, P < 0.005; ***, P < 0.001; §, NS.
In mammals, somatic growth is rapid in embryonic and early postnatal life but decelerates with age. The decline in growth rate is due, in large part, to a decrease in the rate of cell proliferation in several organs. We recently provided evidence in mice that the decline is driven by an extensive genetic program that operates coordinately in many tissues during early postnatal life. This complex program includes the downregulation of several growth-promoting imprinted genes, including Igf2, Plagl1, Mest, Peg3, Dlk1, Gtl2, and Slc38a4. Targeted ablation of the imprinted genes in mice causes reduced body size at birth, indicating that the genes are required for rapid embryonic growth. However, their expression subsequently declines markedly, probably contributing to normal postnatal growth deceleration.
Embryonal cancers are caused by tumors composed of cells that maintain embryonic characteristics and show abnormally rapid proliferation and other malignant features. Such tumors typically present in childhood and include rhabdomyosarcoma, Wilms' tumor, neuroblastoma, and retinoblastoma, which resemble embryonic muscle, renal, neural crest, and retinal tissue, respectively. Some of the most common solid extra-cranial tumors of childhood are the result of embryonal cancers, which account for 8–10% of childhood malignancies.
We hypothesized that failure of the genetic program responsible for normal growth deceleration may contribute to the rapid proliferation and immature characteristics observed in embryonal cancers. Given that the program appears to suppress the rapid cell proliferation that normally occurs in embryonic tissues, we reasoned that failure of the program in a clone of cells would allow persistent rapid proliferation in postnatal life, thus contributing to tumorigenesis.
As a first test of this hypothesis, we focused on the growth-promoting imprinted genes that are normally downregulated postnatally, investigating whether the genes show persistently elevated expression in embryonal cancers. Analysis of available microarray data suggested that some of the imprinted genes, particularly IGF2, PLAGL1, MEST, and PEG3, are overexpressed in some embryonal cancers, especially rhabdomyosarcoma, compared with non-embryonal cancers and with normal tissues. Relative overexpression in rhabdomyosarcoma cell lines compared with non-embryonal cancer cell lines was confirmed by real-time PCR for MEST, PEG3, and IGF2 (Figure 2).
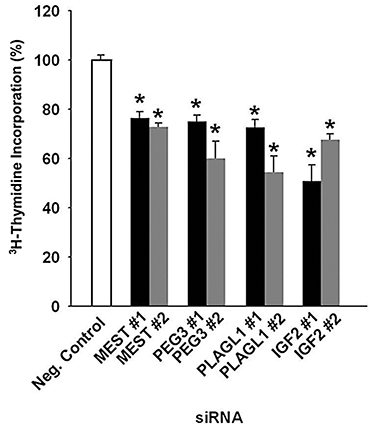
Figure 3. Effects of siRNA transfection on the proliferation of Rh30 rhabdomyosarcoma cells
Cells transfected with siRNA targeting MEST, PEG3, PLAGL1, and IGF2 caused significant reduction in the rate of proliferation when compared with negative control siRNA, as measured by tritiated thymidine incorporation. The effect was confirmed with a second siRNA sequence for each gene target. White bars, transfection with negative control siRNA; black bars, transfection with siRNA #1; grey bars, transfection with siRNA #2. * P < 0.001.
We also found that inhibiting expression of MEST, PEG3, PLAGL1, and IGF2 slowed proliferation of rhabdomyosarcoma cells in culture (Figure 3). For each gene, we knocked down expression using two independent siRNAs and observed that both inhibited proliferation, as assessed by tritiated thymidine incorporation. The results indicate that MEST, PEG3, PLAGL1, and IGF2 are all required for the rapid proliferation observed in these cells and suggest that the observed overexpression of the genes in rhabdomyosarcomas, contributes to their rapid neoplastic proliferation.
Prior studies suggest that MEST, PEG3, PLAGL1, and IGF2 are normally expressed at high levels in embryonic and early postnatal life, promoting the normal rapid body growth that occurs at this time, but are subsequently down-regulated in several tissues simultaneously, contributing to normal somatic growth deceleration. Thus, the current findings suggest that downregulation fails to occur in some embryonal cancers and that this failure contributes to the persistent rapid proliferation of rhabdomyosarcoma cells and possibly their persistent embryonal characteristics.
Previous evidence also suggests that MEST, PEG3, PLAGL1, and IGF2 are members of an imprinted co-regulated gene network. Consistent with this notion, we found that MEST, PEG3, and PLAGL1 expression correlated with IGF2 expression among rhabdomyosarcoma samples. Thus, the data suggest that the genes are regulated coordinately by a common mechanism. Varrault et al. proposed that PLAGL1 may be a master regulator of this imprinted gene network. However, we found that siRNA-mediated knockdown of PLAGL1 expression in a rhabdomyosarcoma cell line did not consistently alter expression of MEST, PEG3, and IGF2. Therefore it is unlikely that this program of declining gene expression is coordinated by PLAGL1 in rhabdomyosarcoma cells. We also tested for other possible interactions among MEST, PEG3, PLAGL1, and IGF2 expression levels, and found that only knockdown of PEG3 reduced expression of PLAGL1. Therefore the findings suggest that the concordant regulation is not coordinated by any of these genes.
In summary, our findings provide evidence that, in addition to IGF2, other imprinted genes, including MEST, PEG3, and PLAGL1, are overexpressed in embryonal tumors including rhabdomyosarcomas and that the overexpression promotes growth of rhabdomyosarcoma cells. Our previous studies suggest that the genes are also highly expressed in embryonic cells and that the subsequent decline in expression contributes to normal growth deceleration. Taken together, the findings suggest that the postnatal program of gene expression that normally limits proliferation in juvenile tissues fails to occur in some embryonal cancers and contributes to the persistent rapid proliferation and perhaps embryonic characteristics of at least one embryonal cancer, rhabdomyosarcoma. Thus, MEST, PEG3, PLAGL1, IGF2, and DLK1 may provide a link between the genetic mechanisms responsible for normal growth deceleration and for the pathologic proliferation of cells in embryonal tumors. Because the imprinted genes that we studied appear to be part of a larger genetic program responsible for growth deceleration, our findings raise the possibility that failure of other components of the program also contribute to the growth of rhabdomyosarcoma and possibly to other embryonal cancers.
Publications
- Lui JC, Nilsson O, Chan Y, Palmer CD, Andrade AC, Hirschhorn J, Baron J. Synthesizing genome-wide association studies and expression microarray reveals novel genes that act in the human growth plate to modulate height. Hum Mol Genet 2012;E-pub ahead of print.
- Rezvani G, Lui JC, Barnes KM, Baron J. A set of imprinted genes required for normal body growth also promotes growth of rhabdomyosarcoma cells. Pediatr Res 2012;71:32-38.
- Dauber A, LaFranchi SH, Maliga Z, Lui C, Moon JE, McDeed C, Henke K, Zonana J, Kingman GA, Pers TH, Baron J, Rosenfeld RG, Hirschhorn JN, Harris MP, Hwa V. Novel microcephalic primordial dwarfism syndrome associated with variants in the centrosomal protein ninein. J Clin Endocrinol Metab 2012;E-pub ahead of print.
- Lui JC, Baron J. Mechanisms limiting body growth in mammals. Endocr Rev 2011;32:422-440.
- Lui JC, Nilsson O, Baron J. Growth plate senescence and catch-up growth. Endocr Dev 2011;21:23-29.
Collaborators
- Andrew Dauber, MD, Boston Children's Hospital, Boston, MA
- Joel Hirschhorn, MD, PhD, Harvard Medical School, Boston, MA
Contact
For further information, contact jeffrey_baron@nih.gov or visit www.nichd.nih.gov/research/atNICHD/Investigators/baron.