

You are here: Home > Molecular Neurophysiology and Biophysics Section
Potassium Channels and Dendritic Function in Hippocampal Pyramidal Neurons

- Dax Hoffman, PhD, Head, Molecular Neurophysiology and Biophysics Section
- Emilie Campanac, PhD, Visiting Fellow
- Erin Gray, PhD, Postdoctoral Fellow
- Eun-young Kim, PhD, Postdoctoral Fellow
- Joshua Lee, BS, Postbaccalaureate Fellow
- Lin Lin, PhD, Microbiologist
- Ying Liu, MD, Biologist
- Laura Long, BA, Postbaccalaureate Fellow
- Ben Throesch, BS, Postbaccalaureate Fellow
The central nervous system underlies all our experiences, actions, emotions, knowledge, and memories. With billions of neurons each firing hundreds of times per second, the complexity of the brain is stunning. To pare down the task of understanding something so complex, our research approach calls for studying the workings of a single central neuron—the pyramidal neuron from the CA1 region of the hippocampus. The hippocampus is essential for long-term memory in humans and is among the first brain regions affected by epilepsy and Alzheimer’s disease. To understand how the hippocampus stores and processes information, we focus on one of its principal cell types, the CA1 pyramidal neuron. Each pyramidal neuron in the CA1 region of the hippocampus receives tens of thousands of inputs onto its dendrites, and it is commonly thought that information is stored by altering the strength of individual synapses (synaptic plasticity). Recent evidence suggests that the regulation of synaptic surface expression of glutamate receptors can, in part, determine synaptic strength. However, the dendrites contain an abundance of ion channels that are involved in receiving, transforming, and relaying information in the dendrites, adding an additional layer of complexity to neuronal information processing.
We found that the A-type potassium channel subunit Kv4.2 is highly expressed in the dendritic regions of CA1 neurons in the hippocampus and, as one of the primary regulators of dendritic excitability, plays a pivotal role in information processing. Kv4.2 is targeted for modulation during the types of plasticity thought to underlie learning and memory. Moreover, we found that the functional expression level of Kv4.2 regulates the subtype expression of NMDA–type glutamate receptors, the predominant molecular devices controlling synaptic plasticity and memory. We are currently following up on these findings with more detailed investigations into the mechanisms of activity-dependent Kv4.2 regulation and its role in neuronal development. In addition, we are investigating the role of dendritic voltage-gated channels in CNS disorders, including autism-spectrum disorder and Alzheimer’s disease.
Voltage-gated ion channel expression and trafficking in dendrites
Kv4.2 control of firing patterns in hippocampal CA1 pyramidal neurons
Although recent molecular cloning studies found that several families of voltage-gated K+ channel genes are expressed in the mammalian brain, information regarding the relationship between the protein products of these genes and their various neuronal functions is still lacking. Our lab used a combination of molecular, electrophysiological, and imaging techniques to show that Kv4.2, an A-type voltage-gated potassium channel subunit, controls action potential (AP) half-width, frequency-dependent AP broadening, and dendritic AP propagation. More recently, we examined the role of A-type K+ channels in regulating synaptic plasticity, neuronal development and disease (1).
Kv4.2 trafficking in CA1 pyramidal neuron dendrites
We previously reported that neuronal stimulation results in a redistribution of Kv4.2 channels away from dendritic spines to the dendritic shaft. Such activity-dependent redistribution of Kv4.2 requires activation of NMDA-type glutamate receptors and calcium influx, two requirements shared with synaptic plasticity, which is thought to underlie learning and memory. Given the nonuniform distribution of Kv4.2 channels in CA1 dendrites, former postdoctoral fellow Mike Nestor performed experiments to test the hypothesis that Kv4.2 channels are differentially trafficked at different regions along the apical dendrite during basal activity and upon stimulation in CA1 neurons (2). Proximal (50–150 μm from the soma, primary and oblique) and distal (more than 200 μm from the soma) apical dendrites were selected. He used the fluorescence recovery after photobleaching (FRAP) technique to measure basal cycling rates of EGFP-tagged Kv4.2 (Kv4.2g). We found that the cycling rate of Kv4.2 channels was one order of magnitude slower at both primary and oblique dendrites between 50-150 μm from the soma. Kv4.2 channel cycling increased significantly at 200-250 μm from the soma. Expression of a Kv4.2 mutant lacking a phosphorylation site for protein kinase-A (Kv4.2gS552A) abolished this distance-dependent change in channel cycling, demonstrating that phosphorylation by PKA underlies the increased mobility in distal dendrites. These results indicate that distance-dependent Kv4.2 mobility is regulated by activity-dependent phosphorylation of Kv4.2 by PKA. Josh Lee has followed up on this project, aiming to uncover the pathway of internalized Kv4.2 as well as proteins that may be involved in directing its path. To date, he has shown that ubiquitin and the ubiquitin ligase Nedd4-2 interact with Kv4.2 when overexpressed in a non-neuronal cell line. In the same cell line, he showed that ubiquitin facilitates degradation of Kv4.2 compared with the basal rate of degradation. We are simultaneously conducting experiments to visually track the movement of Kv4.2. Colocalizing Kv4.2 with markers of well characterized endosomal compartments will reveal its pathway.
In a collaboration with the Juan Bonifacino lab, Laura Long is examining the interaction of Kv4.2 with the clathrin-associated adaptor protein complex AP-1A. AP-1A is responsible for intracellular trafficking and has recently been implicated in the determination of receptor polarity in neurons. Molecular approaches confirmed that Kv4.2 does indeed interact with AP-1A, and coimmunoprecipitation, and yeast in situ hybridization assays suggest that this binding occurs at both the N-terminal and C-terminal tails of Kv4.2. Long is currently investigating the functional implications of this interaction through imaging and shRNA knockouts.
Voltage-gated calcium channel trafficking
Erin Gray began a research project investigating activity-dependent trafficking of another important dendritically localized channel, the voltage-gated calcium channel Cav2.3. In dendritic spines, Cav2.3 selectively generates large calcium spikes, which shape both synaptic potentials and back-propagating action potentials. Furthermore, modulation of these channels by activation of kinases such as PKC and CamKII can dramatically affect synaptic plasticity and neuronal excitability. Gray cloned Cav2.3 into expression plasmids to generate Cav2.3 with a GFP tag on the channel's N- or C-terminus. After confirming the functional expression of all three constructs in HEK cells, she expressed the recombinant channels in dissociated hippocampal neuron culture. She will begin imaging studies to determine the localization and activity-dependent trafficking of Cav2.3 in neurons.
Role of voltage-gated ion channels in synaptic development and disease
Role of Kv4.2 channels in synaptic plasticity and development
We have found that altering functional Kv4.2 expression level leads to a rapid, bidirectional remodeling of CA1 synapses. Neurons exhibiting enhanced A-type K+ current (IA) show a decrease in relative synaptic NR2B/NR2A subunit composition and do not exhibit the form of synaptic plasticity known as long-term potentiation or LTP. Conversely, reducing IA by expression of a Kv4.2 dominant negative or through genomic knockout of Kv4.2 led to an increased fraction of synaptic NR2B/NR2A and enhanced LTP. Our data suggest that A-type K+ channels are an integral part of a synaptic complex that regulates Ca2+ signaling through spontaneous NMDA receptor activation to control synaptic NMDA receptor expression and plasticity. Additional advances made by Eun-young Kim included an investigation into the role of Kv4.2 in controlling the expression of synaptic NMDA receptors in vivo and during development. The synaptic NR2B fraction is developmentally regulated, with implications for synaptic plasticity and learning and memory as well as diseases associated with learning impairments. Kim published a 2013 Journal of Neuroscience paper, in which she reported that in vivo injection of virus to alter Kv4.2 expression levels bidirectionally regulates NR2B subunit expression throughout development (3).
Role of the Kv4.2 auxiliary subunit DPP6 in synaptic plasticity and development
Studies in heterologous expression systems have shown that Kv4.2 subunits interact with transmembrane DPP6 proteins to regulate channel trafficking and properties. The DPP6 auxiliary subunit protein, which is expressed in CA1 neurons, has recently been identified in large copy-number variants screens from some populations as an Autism Spectrum Disorder and amyotropic lateral sclerosis (ALS) target gene. DPP6 enhances the opening probability of Kv4 channels and increases channel surface expression in heterologous systems. In dendritic recordings from DPP6 knockout mice, former graduate student Wei Sun discovered that DPP6 is critical for generating the A-type K+ current gradient observed in CA1 dendrites. The loss this gradient led to hyper-excitable dendrites, with implications for information storage and coding.
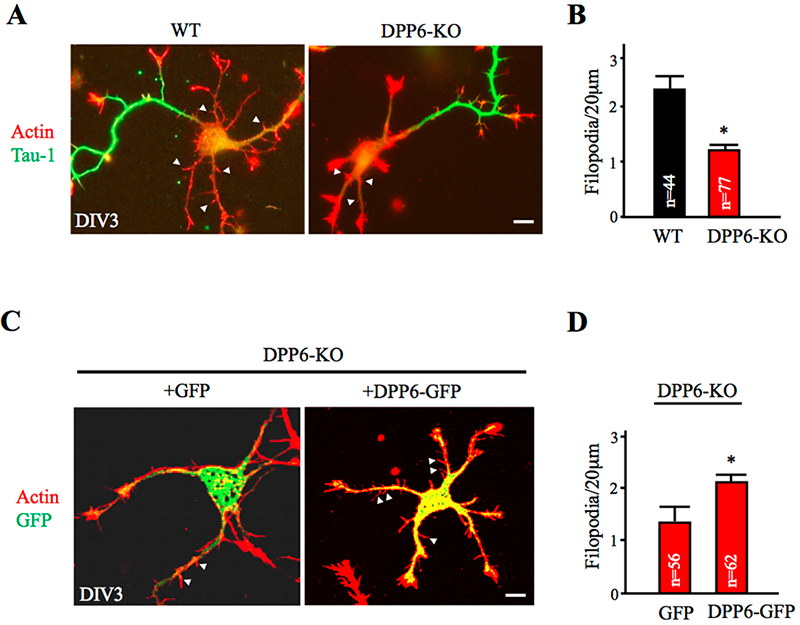
This year, Lin Lin reported in a Nature Communications paper that knockdown and genetic deletion of DPP6 reveals its importance for the formation and stability of dendritic filopodia during early neuronal development (Figure 1 and reference 4). Additionally, hippocampal neurons lacking DPP6 exhibited a sparser dendritic branching pattern and fewer spines throughout development and into adulthood. In electrophysiological and imaging experiments, we showed that these deficits lead to fewer functional synapses and occur independently of the potassium channel subunit Kv4.2. We found that the extracellular domain of DPP6 interacts with a filopodia-associated myosin as well as with fibronectin in the extracellular matrix. DPP6 therefore plays an unexpected but important role in cell adhesion and motility, impacting hippocampal synaptic development and function.
Click image to enlarge.
Figure 1. Dendritic filopodia formation is reduced in DPP6-KO hippocampal neurons and rescued by DPP6 expression.
(A,B) DIV3 WT or DPP6-KO hippocampal neurons visualized for actin filaments with TRITC-phalloidin (1:1000, Sigma) and for axons with marker Tau-1 (1:1000, Millipore). The density of immature dendritic filopodia (arrowheads) is reduced in the DPP6-KO neurons (n=77) compared to the WT (n=44). (C,D) DIV3 DPP6-KO hippocampal neurons expressing either GFP control or DPP6-GFP, cultured for three days, and visualized for actin filaments with TRITC-phalloidin. The density of filopodia is elevated in the DPP6-KO neurons that express DPP6-GFP (n=62) compared with the GFP controls (n=56). Scale bars, 10μm. Error bars represent the mean±SEM; *p< 0.05.
Dendritic intrinsic excitability changes in disease
Intrinsic excitability changes have been observed upon drug addiction. The psychostimulant effects of the addictive drug cocaine are attributed to inhibition of the dopamine transporter, which increases dopaminergic transmission. Chronic exposure to cocaine leads to neurodaptations in several membrane conductances of neurons localized in the medial prefrontal cortex (mPFC) and nucleus accumbens. In a Journal of Neurophysiology paper published this year, Emily Campanac reported that repeated cocaine exposure increases fast-spiking interneuron excitability in the rat mPFC (5). After cocaine withdrawal, interneurons showed an increase in action potential firing, increased input resistance, and decreased hyperpolarization-activated current. We also observed a reduction in miniature excitatory postsynaptic currents, whereas miniature inhibitory postsynaptic current activity was unaffected. In animals with a cocaine history, dopamine receptor D(2) activation was less effective in increasing interneuron intrinsic excitability. Interestingly, these alterations were only manifested one week or more after the last cocaine exposure. This suggests that the dampening of D(2)-receptor–mediated response may be a compensatory mechanism to rein in the excitability of interneurons.
Neuronal hyperexcitability is an early feature of Alzheimer’s disease. The underlying cellular mechanisms are unclear however. Ben Throesch examined dendritic excitability directly by patch-clamp recordings on dendrites of hippocampal neurons in mice expressing elevated levels of amyloid-β. In a paper in preparation, he describes his results showing that neuronal dendrites were hyper-excitable due to a decrease in Kv4.2 expression, while the soma showed normal firing.
Additional Funding
- National Institute of Child Health and Human Development Scientific Director’s Award (2012)
Publications
- Nestor MW, Hoffman DA. Aberrant dendritic excitability: a common pathophysiology in CNS disorders affecting memory? Mol Neurobiol 2012;45:478-487.
- Nestor MW, Hoffman DA. Differential cycling rates of Kv4.2 channels in proximal and distal dendrites of hippocampal CA1 pyramidal neurons. Hippocampus 2012;22:969-980.
- Kim E-Y, Hoffman DA. Dynamic regulation of synaptic maturation state by voltage-gated A-type K+ channels in CA1 hippocampal pyramidal neurons. J Neurosci 2012;32:14427-14432.
- Lin L, Sun W, Throesch B, Kung F, Decoster JT, Berner CJ, Cheney RE, Rudy B, Hoffman DA. DPP6 regulation of dendritic morphogenesis impacts hippocampal synaptic development. Nat Commun 2013;4:2270.
- Campanac E, Hoffman DA. Repeated cocaine exposure increases fast-spiking interneuron excitability in the rat medial prefrontal cortex. J Neurophysiol 2013;109:2781-2792.
Contact
For more information, email hoffmand@mail.nih.gov or visit neuroscience.nih.gov/Faculty/Profile/dax-hoffman.aspx.