

You are here: Home > Section on Intracellular Protein Trafficking
Protein Sorting in the Endosomal-Lysosomal System

- Juan S. Bonifacino, PhD, Head, Section on Intracellular Protein Trafficking
- Rafael Mattera, PhD, Staff Scientist
- Xiaolin Zhu, RN, Technician
- Yu Chen, PhD, Research Fellow
- Loreto Cuitiño, PhD, Visiting Fellow
- Ginny Farias, PhD, Visiting Fellow
- David Gershlick, PhD, Visiting Fellow
- Xiaoli Guo, PhD, Visiting Fellow
- Shweta Jain, PhD, Visiting Fellow
- Rui Jia, PhD, Visiting Fellow
- Sang-Yoon Park, PhD, Visiting Fellow
- Jing Pu, PhD, Visiting Fellow
- Stine C. Klinger, PhD, Guest Researcher
Click image to enlarge.
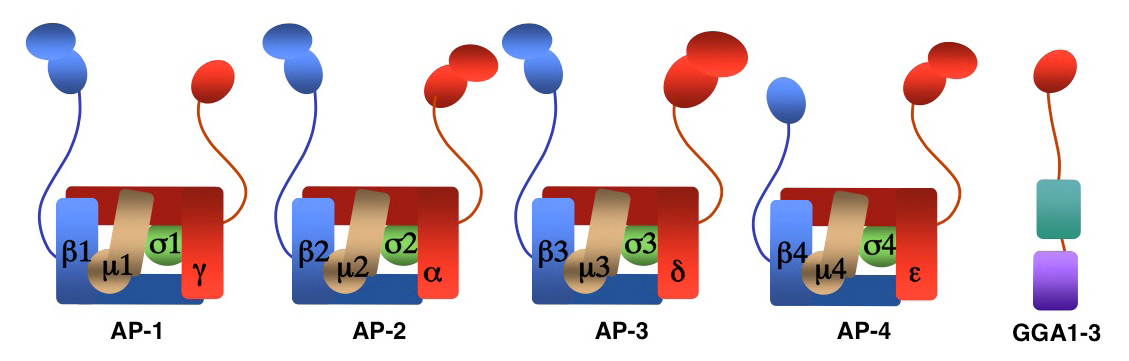
Figure 1. Structure of AP complexes and GGAs
We investigate the molecular mechanisms by which transmembrane proteins are sorted to various compartments of the endomembrane system such as endosomes, lysosomes, lysosome-related organelles (e.g., melanosomes and platelet dense bodies) and specific domains of the plasma membrane in polarized cells (e.g., epithelial cells and neurons). Sorting is often mediated by recognition of signals present in the cytosolic domains of the transmembrane proteins by adaptor proteins that are components of membrane coats (e.g., clathrin coats). Among these adaptor proteins are the heterotetrameric AP-1, AP-2, AP-3, and AP-4 complexes, the monomeric GGA proteins, and the heteroligomeric retromer complex. Sorting also involves the function of other components of the trafficking machinery that mediate vesicle-tethering and fusion, such as the multisubunit tethering complex GARP and cognate SNARE proteins. Current work in our laboratory aims to elucidate the structure, regulation, and physiological roles of coat proteins and vesicle tethering factors, as well as to investigate human diseases that result from genetic defects of these proteins (e.g., Hermansky-Pudlak syndrome; neurodegenerative and neurodevelopmental disorders) or their exploitation by pathogens (e.g., HIV-1).
An AP-1/clathrin pathway for the sorting of transmembrane receptors to the somatodendritic domain of hippocampal neurons
Click image to enlarge.
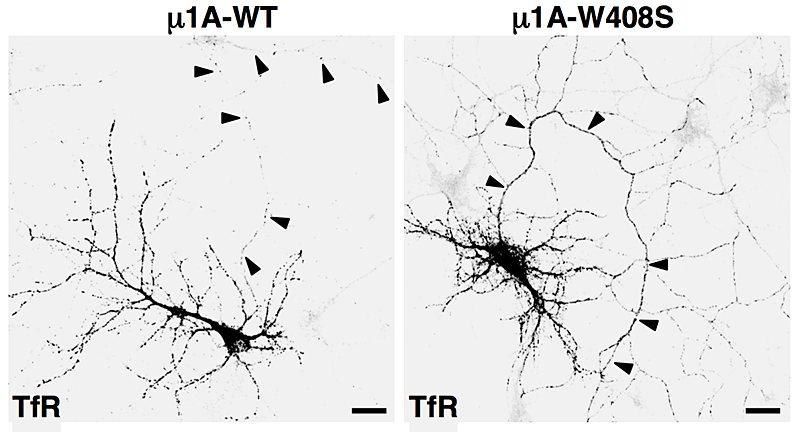
Figure 2. Overexpression of dominant-negative mutant of AP-1 subunit (μ1A-W408S) causes missorting of transferrin receptor (TfR) to the axon (arrowheads).
AP-1, AP-2, AP-3, and AP-4 are adaptor protein complexes that recognize two types of sorting signal, referred to as tyrosine-based and dileucine-based signals. Previous studies from our laboratory showed that tyrosine-based signals bind to the mu1, mu2, and mu3 subunits, whereas dileucine-based signals bind to a combination (i.e., a hemicomplex) of two subunits, gamma-sigma1, alpha-sigma2, and delta-sigma3, from the corresponding AP-1, AP-2, and AP-3 complexes. To date, the AP-4 complex has only been shown to recognize tyrosine-based signals via its mu4 subunit. A major goal of our recent work has been the analysis of the role of signal-adaptor interactions in polarized sorting in neurons. Neurons are polarized into dendrites, soma, and axons. The plasma membrane of each of these domains possesses a distinct set of transmembrane proteins, including receptors, channels, transporters, and adhesion molecules. We hypothesized that sorting to these domains was mediated by interaction of sorting signals with AP complexes. Our studies showed that the cytosolic tails of various transmembrane receptors, including the transferrin receptor (TfR), the Coxsackie virus and adenovirus receptor (CAR), and the glutamate receptor proteins mGluR1, NR2A, and NR2B, all have information leading to the sorting of the proteins to the somatodendritic domain of hippocampal neurons. In the case of TfR and CAR, the information resides in tyrosine-based sorting signals. Protein interaction analyses showed that the tails of the receptors bind to the mu1A subunit of AP-1. Dominant-negative interference and RNAi approaches demonstrated that interaction of their cytosolic tails with AP-1 was responsible for somatodendritic sorting. Sorting involved exclusion of the receptor proteins from vesicular transport carriers destined for the axonal domain at the level of the soma. Interference with AP-1–dependent somatodendritic sorting caused defective maturation of dendritic spines and reduced the number of excitatory synapses.
Co-assembly of viral envelope glycoproteins regulates their polarized sorting in neurons.
We recently extended our studies on the mechanisms of polarized sorting in neurons to the attachment (NiV-G) and fusion (NiV-F) glycoproteins of Nipah virus, a neuroinvasive pathogen that causes fatal human encephalitis. When analyzed individually, NiV-G is delivered to both the axonal and somatodendritic domains (i.e., it is non-polarized). In contrast, NiV-F is exclusively targeted to the somatodendritic domain by virtue of the interaction of a tyrosine-based sorting signal with the mu1A subunit of AP-1. Importantly, co-expression with NiV-G causes NiV-F to lose its somatodendritic polarity, becoming evenly distributed between the somatodendritic and axonal domains. The redistribution is attributable to the incorporation of NiV-F into axonal transport carriers in the presence of NiV-G. We also observed that NiV-F exhibits a faster rate of biosynthetic transport than does NiV-G. The findings led us to propose that coordinated interactions of viral glycoproteins with the host’s sorting machinery and between themselves allow temporal and spatial regulation of the glycoproteins' distribution between the somatodendritic and axonal domains of neurons, a process that may have implications for viral spread through synaptic contacts.
Polarized sorting of the copper transporter ATP7B in neurons is mediated by recognition of a dileucine-based signal by AP-1.
As mentioned above, AP-1 also binds to dileucine-based signals by virtue of interactions with a site on the gamma-sigma1 hemicomplex. We recently found that interactions of AP-1 with dileucine-based signals in the copper transporter ATP7B and the vesicle-SNARE VAMP4 also mediate sorting to the somatodendritic domain of rat hippocampal neurons. ATP7B localizes to the trans-Golgi network (TGN) under low-copper conditions but redistributes to the cell periphery under high-copper conditions in hippocampal neurons. Furthermore, under low-copper conditions, ATP7B is restricted to the soma and dendrites and excluded from the axon. Such somatodendritic polarity is lost upon mutation of the dileucine-based signal in ATP7B or overexpression of a dominant-negative sigma1 mutant incapable of binding to such signals. High copper levels also cause loss of somatodendritic polarity. The findings indicate that copper levels regulate not only the distribution of ATP7B between the TGN and peripheral vesicles but also the polarized sorting of the transporter between the somatodendritic and axonal domains.
Mu1 subunit isoforms expand the repertoire of basolateral sorting signal recognition in epithelial cells.
Some AP-1 subunits occur as various isoforms encoded by different genes. For example, there are two mu1 subunit isoforms (mu1A and mu1B) and three sigma1 subunit isoforms (sigma1A, sigma1B, and sigma1C). We investigated the physiologic significance for the existence of these isoforms. Years ago, we found that mu1A is ubiquitously expressed, whereas mu1B is exclusively expressed in epithelial cells. Using advanced confocal microscopy techniques, including spinning disk, total internal reflection fluorescence (TIRF) microscopy, and super-resolution structured illumination microscopy, we found that mu1A and mu1B similarly localize to both the TGN and endosomes. However, each isoform exhibits preferences for a distinct set of signals. For example, yeast two-hybrid and glutathione S-transferase (GST) pull-down experiments showed that the non-canonical tyrosine-based signals and the clusters of acidic residues that mediate basolateral sorting of the low-density lipoprotein receptor preferentially bind to mu1B. We conclude that the existence of mu1 subunit isoforms expands the repertoire of AP-1 signal recognition in epithelial cells, allowing for efficient and regulated sorting of a broader set of cargoes to the basolateral surface. Taken together, our studies in neurons and epithelial cells establish the AP-1 complex as a global regulator of polarized sorting.
Defects in polarized sorting may underlie the pathogenesis of neurological/cutaneous disorders caused by mutation in sigma1 subunit isoforms.
Recently, mutations in each of the three sigma1 subunit isoforms in humans were shown to cause neurodevelopmental and/or cutaneous disorders such as the MEDNIK syndrome (sigma1A), Fried/Pettigrew syndrome (sigma1B), and pustular psoriasis (sigma1C). We had previously found that sigma1 isoforms recognize overlapping but distinct sets of dileucine-based signals. Thus, diseases caused by sigma1 isoform mutations could result from missorting of specific sets of transmembrane proteins bearing dileucine-based signals. In the case of the MEDNIK syndrome, failure of polarized sorting of ATP7B in the liver and brain could contribute to the copper metabolism defects that underlie some of the symptoms of this disease.
Structural insights into the mechanisms of vesicle tethering and fusion
We also made progress in elucidating the molecular mechanisms of retrograde transport from endosomes to the TGN. In collaboration with Aitor Hierro, we uncovered the structural mechanism by which the tethering factor GARP interacts with its cognate vesicle fusion SNARE proteins. X-ray crystallographic analyses showed how the Ang2 subunit of GARP binds to the Habc domain of the Syntaxin 6 SNARE. The findings highlight a key event in the pathway by which transport vesicles emanating from endosomes fuse with the TGN. The integrity of the pathway is essential for neuronal function and viability, as demonstrated by the discovery that mutations in the Vps53 subunit of GARP cause progressive cerebello-cerebral atrophy type 2 (PCCA2).
The Nef and Vpu accessory proteins of HIV-1
Primate immunodeficiency viruses target helper T-cells and macrophages/monocytes through binding of the viral envelope glycoprotein to a combination of CD4 and a chemokine receptor (CCR4 or CXCR5) on the surface of the host cells. Strikingly, infection results in rapid and sustained downregulation of CD4 and, to a lesser extent, the chemokine receptors. Downregulation of these viral co-receptors prevents superinfection, promotes virion release and interferes with the immune response, leading to the establishment of a robust infection. CD4 downregulation is so important to the life cycle of human immunodeficiency virus-1 (HIV-1) that two accessory proteins, Nef and Vpu, encoded in the viral genome, are devoted to the task. Indeed, Nef and Vpu are critical for the progression from infection to AIDS, a fact that is best illustrated by the existence of long-term non-progressors who are infected with HIV-1 strains bearing inactivating mutations in the genes encoding these proteins. Therefore, pharmacologic or biologic perturbation of Nef and/or Vpu has the potential to prevent the pathogenic effects of HIV-1. To date, however, this potential has not been realized, mainly because Nef and Vpu have no enzymatic activity and their mechanisms of action are insufficiently understood.
Structural basis for CD4 downregulation by the Nef protein of HIV-1
In previous work, we made substantial progress towards elucidating the mechanism of CD4 downregulation by Nef. We found that Nef connects surface CD4 to both the endocytic and lysosomal targeting machineries, leading to efficient and sustained removal of CD4 from the host cells early during infection. We discovered that the role of Nef in CD4 internalization involves interaction with the AP-2 clathrin adaptor. A dileucine motif and a diacidic motif in a central loop of Nef were found to be essential for interaction with a site on the AP-2 alpha-sigma2 hemicomplex and for CD4 downregulation, but the structural details of these interactions were not known. In collaboration with James Hurley, we solved the crystal structure of Nef bound to the alpha and sigma2 subunits of AP-2. The structure revealed that the Nef dileucine motif directly interacts with a binding site for host dileucine signal–containing cargo proteins on alpha-sigma2. The Nef diacidic motif, on the other hand, does not directly interact with AP-2 but stabilizes a binding-competent conformation of the central loop. In addition, the structure showed that the Nef core is involved in direct contacts with alpha-sigma2 while also serving as a scaffold to position the central loop. In conjunction with AP-2 binding and CD4 downregulation analyses, mutagenesis confirmed the importance of the residues identified in the crystal structure. The newly discovered interfaces revealed by these analyses are not known to be used by any host-cell transmembrane proteins and may therefore be specific for Nef. If so, they may represent an “Achilles heel” that could be exploited for the development of novel anti-Nef agents.
The host-cell protein Alix/AIP1 is required for targeting of internalized CD4 to the multivesicular body pathway by Nef.
Subsequent to induction of CD4 internalization by an AP-2/clathrin pathway, Nef promotes delivery of internalized CD4 to the multivesicular body pathway (MVB) for eventual degradation in lysosomes. In previous work, we found that CD4 targeting to the MVB pathway was independent of CD4 ubiquitination. In collaboration with Luis daSilva, we recently found that the targeting depends on direct interaction of Nef with Alix/AIP1, a protein associated with the Endosomal Sorting Complexes Required for Transport (ESCRT) machinery that assists with cargo recruitment and intraluminal vesicle formation in MVBs. We showed that Nef interacts with both the Bro1 and V domains of Alix. Depletion of Alix or overexpression of the Alix V domain impaired lysosomal degradation of CD4 induced by Nef. In contrast, V-domain overexpression did not prevent cell-surface removal of CD4 by Nef or protein targeting to the canonical, ubiquitination-dependent MVB pathway. We also showed that the Nef-Alix interaction occurs in late endosomes that are enriched in internalized CD4. Together, the results indicated that Alix functions as an adaptor for the ESCRT–dependent, ubiquitin-independent targeting of CD4 to the MVB pathway induced by Nef.
Vpu induces caspase-mediated cleavage of the transcription factor IRF3.
We also sought to elucidate the mechanisms by which Vpu interferes with host-cell factors other than CD4. One of these is the Interferon Regulatory Factor 3 (IRF3), a key transcription factor in the regulation of the host innate immune response to viral infection. IRF3 activation induces the expression of type I interferons as well as several interferon-stimulated genes. The IRF3 gene encodes a 427–amino acid protein containing a DNA–binding domain (residues 1–110), an IRF–association domain (residues 198–374), and a transactivation domain (residues 134–394). Because HIV-1 infection does not induce expression of type I interferons, it is has been suggested that it must have mechanisms for antagonizing IRF3 function. HIV-1 Vpu was thought to promote lysosomal degradation of IRF3. However, in collaboration with Abdul Waheed and Eric Freed, we found that Vpu does not induce lysosomal degradation of IRF3, but causes caspase-mediated cleavage of IRF3 at residue Asp121, resulting in removal of the DNA–binding domain from the rest of the protein. The cleavage was observed both in non–T cells transfected with a plasmid encoding Vpu and in Jurkat T cells infected with a VSV-G–pseudotyped HIV-1. Two other HIV-1 accessory proteins, Vif and Vpr, also contribute to the induction of IRF3 cleavage in both the transfection and infection systems. The C-terminal IRF3 fragment interferes with the transcriptional activity of full-length IRF3. Cleavage of IRF3 under all these conditions correlates with cleavage of poly(ADP-ribose) polymerase, an indicator of apoptosis. We concluded that Vpu attenuates the anti-viral response by partially inactivating IRF3 while host cells undergo apoptosis.
Additional Funding
- Intramural AIDS Targeted Antiviral Program (IATAP)
Publications
- Farías GG, Cuitino L, Guo X, Ren X, Jarnik M, Mattera R, Bonifacino JS. Signal-mediated, AP-1/clathrin-dependent sorting of transmembrane receptors to the somatodendritic domain of hippocampal neurons. Neuron 2012;75:810-823.
- Ren X, Farías GG, Canagarajah BJ, Bonifacino JS, Hurley JH. Structural basis for recruitment and activation of the AP-1 clathrin adaptor complex by Arf1. Cell 2013;152:755-767.
- Mardones GA, Burgos PV, Lin Y, Kloer DP, Magadán JG, Hurley JH, Bonifacino JS. Structural basis for the recognition of tyrosine-based sorting signals by the mu3A subunit of the AP-3 adaptor complex. J Biol Chem 2013;288:9563-9571.
- Zhang ZR, Bonifacino JS, Hegde R. Deubiquitinases sharpen substrate discrimination during membrane protein degradation from the ER. Cell 2013;164:609-622.
- Abascal-Palacios G, Schindler C, Rojas A, Bonifacino JS, Hierro A. Structural basis for the interaction of the Golgi-Associated Retrograde Protein (GARP) complex with the t-SNARE Syntaxin 6. Structure 2013;21:1698-1706.
Collaborators
- Luis L. DaSilva, PhD, University of São Paulo, São Paulo, Brazil
- Eric O. Freed, PhD, HIV Drug Resistance Program, Center for Cancer Research, NCI, Frederick, MD
- Aitor Hierro, PhD, CIC-bioGUNE, Bilbao, Spain
- James Hurley, PhD, Laboratory of Molecular Biology, NIDDK, Bethesda, MD (now at now at UC Berkeley, CA)
- Abdul Waheed, PhD, Retroviral Replication Laboratory, Center for Cancer Research, NCI, Frederick, MD
Contact
For further information, contact juan@helix.nih.gov or visit cbmp.nichd.nih.gov/sipt.