

You are here: Home > Section on Organelle Biology
Interplay between Membrane Organelles, Cytoskeleton, and Metabolism in Cell Organization and Function

- Jennifer Lippincott-Schwartz, PhD, Head, Section on Organelle Biology
- Yu Chen, PhD, Visiting Fellow
- Sarah Cohen, PhD, Visiting Fellow
- Uri Manor, PhD, Postdoctoral Fellow
- Carolyn Ott, PhD, Postdoctoral Fellow
- Christopher Obara, PhD, Postdoctoral Fellow
- Timothy Petri, PhD, Postdoctoral Fellow
- Prabuddha Sengupta, PhD, Postdoctoral Fellow
- Arnold Seo, PhD, Postdoctoral Fellow
- Alex Valm, PhD, Postdoctoral Fellow
- Aubrey Weigel, PhD, Postdoctoral Fellow
- Lingfeng Chen, PhD, Volunteer
- Prasanna Satpute, PhD, Volunteer
- Alex Ritter, BA, Graduate Student
- Bennett Waxse, BA, Graduate student
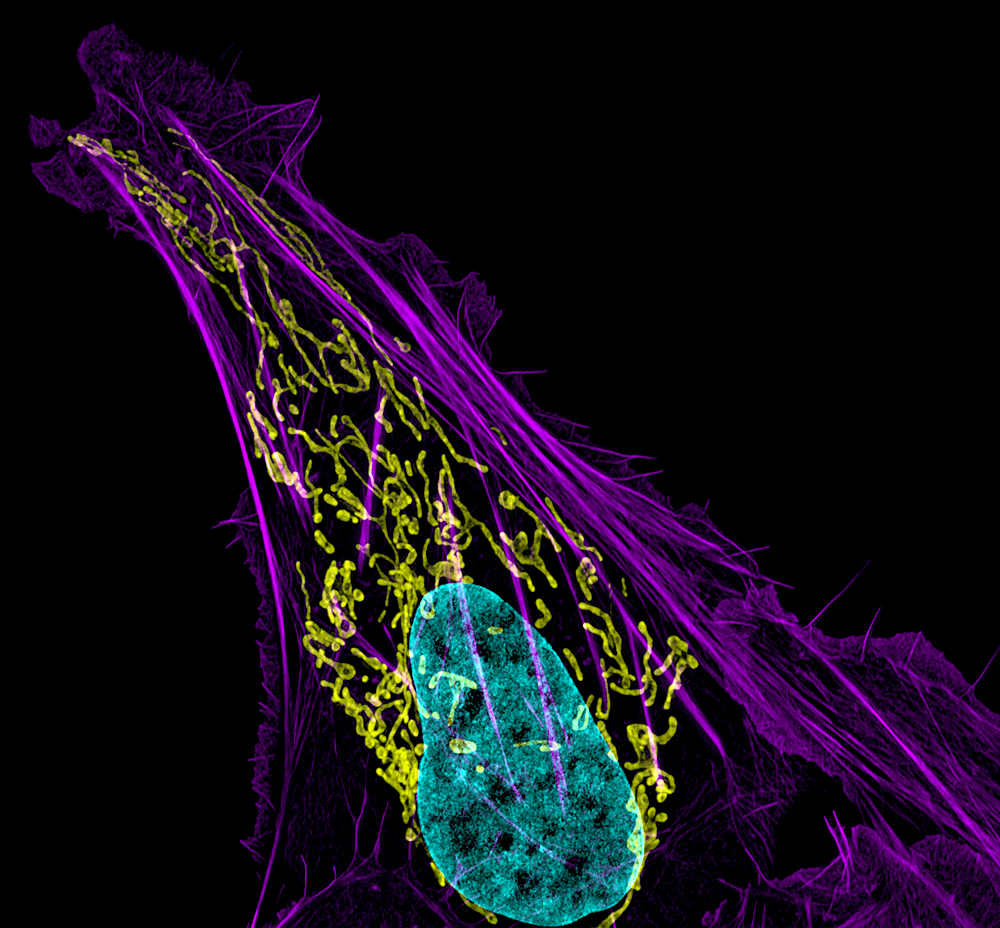
Click image to enlarge.
Figure 1. Mitochondrial and actin organization in a crawling cell
Structured illumination microscope (SIM) image of a U2OS (osteoblastoma) cell showing mitochondria stained with MITO-FRP (pink), actin stained with Alexa Fluor 488® phalloidin (green), and DNA stained with Hoechst 33342 (blue)
We investigate the global principles underlying cell behavior at both small and large spatial scales. At the small scale, we employ the super-resolution imaging techniques of photoactivated localization microscopy (PALM), interferometric 3D PALM, single-particle tracking PALM, and pair-correlation PALM to map the spatial organization, stoichiometry, and dynamics of proteins associated with various membrane-bound compartments and with the cytoskeleton. We also employ fluorescence photobleaching, photoactivation, fluorescence correlation, and fluorescence energy transfer methods to measure protein-protein interactions, protein turnover rates, and protein association rates. Such approaches allow us to assay cellular functions, including receptor stoichiometry and protein clustering and diffusion behavior at the nanometric scale in living cells. At the large scale, we investigate how complex behaviors of cells arise, such as cell crawling, polarization, furrowing, cytokinesis, cell fate determination, viral budding, and intercellular transfer. We study these complex behaviors by quantitatively analyzing diverse intracellular processes, including membrane trafficking, autophagy, actin/microtubule dynamics, and organelle assembly/disassembly pathways, which undergo dramatic changes as cells alter their behavior and organization throughout life. To assist these efforts, we combine various fluorescence-based imaging approaches, including total internal reflection fluorescence (TIRF) microscopy imaging and spinning-disk and laser-scanning confocal microscopy, with FRAP (fluorescence recovery after photobleaching), FLIP (fluorescence loss in photobleaching), and photoactivation to obtain large image data sets. We process the data sets computationally to extract biochemical and biophysical parameters, which can be related to results from conventional biochemical assays. We then use the results to generate mechanistic understanding and predictive models of the behavior of cells and subcellular structures (including ER, Golgi, cilia, endosomes, lysosomes, autophagosomes, and mitochondria) under healthy and pathological conditions.
Cell shape control by a counterbalanced adhesion/contraction mechanism
We combined 3D super-resolution analyses of crawling cells with the development of a biophysical modeling scheme to show that the seemingly complex process of lamella flattening in the crawling cell can be explained based on mechanical principles and cytoskeletal reorganization. Live-cell microscopy data showed a crucial role for a contractile meshwork at the top of the cell, which is composed of actin arcs and myosin IIA filaments. We found the contractile actin meshwork to be organized like muscle sarcomeres, with repeating myosin II filaments separated by the actin-bundling protein α-actinin, and to be mechanically coupled to noncontractile dorsal actin fibers that run from top to bottom in the cell. When the meshwork contracted, it pulled the dorsal fibers away from the substrate. The pulling force was counterbalanced by the dorsal fibers' attachment to focal adhesions, causing the fibers to bend downward and flattening the cell. This model of cell-shape control is likely to be relevant for understanding how cells configure themselves to complex surfaces, protrude into tight spaces, and generate three-dimensional forces on the growth substrate under both healthy and diseased conditions.
Nanoscale organization of ESCRT fission machinery during HIV virus budding
Key host factors on which the human immunodeficiency virus (HIV) relies to complete its infection cycle are the endosomal sorting complexes required for transport (ESCRT). By recruiting this machinery, HIV is able to mediate the final step of virus-particle fission from the membrane. Otherwise, virus egress is severely inhibited. Despite great progress in demonstrating the ESCRT machinery’s role in mediating HIV abscission, the nanoscale organization, and thus function, of ESCRT subcomplexes at native HIV assembly sites remains poorly defined. We applied three-dimensional super-resolution microscopy and correlative electron microscopy to delineate the organization of ESCRT components at HIV assembly sites. We observed ESCRT subunits localized within the head of budding virions and released particles, with head-localized levels of the component CHMP2A (charged multivesicular body protein 2A) declining relative to the components Tsg101 and CHMP4B upon virus abscission. Thus, the driving force for HIV release may derive from initial scaffolding of ESCRT subunits within the viral bud interior followed by plasma-membrane association and selective remodeling of ESCRT subunits.
ER stress–dependent clearance of misfolded proteins via the secretory pathway

Click image to enlarge.
Figure 2. Mitochondria morphology in NRK cells
Image, obtained by structured illumination microscopy, of mitochondria labeled with GFP-tagged prohibitin
Proteins destined for the cell surface are first assessed in the endoplasmic reticulum (ER) for proper folding before release into the secretory pathway. This ensures that defective proteins are prevented from entering the extracellular environment, where they could be disruptive. We reported that, when ER–folding capacity is saturated during stress, misfolded glycosylphosphatidylinositol-anchored proteins dissociate from resident ER chaperones, engage export receptors, and quantitatively leave the ER via vesicular transport to the Golgi. Clearance from the ER commences within minutes of acute ER stress, before the transcriptional component of the unfolded-protein response is activated. The aberrant proteins then access the cell surface transiently before destruction in lysosomes. Inhibiting this stress-induced pathway by depleting the ER–export receptors leads to aggregation of the ER–retained misfolded protein. The rapid response thus alleviates the elevated burden of misfolded proteins in the ER at the onset of ER stress, promoting protein homeostasis in the ER.
MicroRNA binding to the HIV-1 Gag protein inhibits Gag assembly and virus production.
MicroRNAs (miRNAs) are small, 18–22 nt long, noncoding RNAs that act as potent negative gene regulators in a variety of physiological and pathological processes. To repress gene expression, miRNAs are packaged into RNA–induced silencing complexes (RISCs), which target mRNAs for degradation and/or translational repression in a sequence-specific manner. Recently, miRNAs have been shown to also interact with proteins outside RISCs, impacting cellular processes through mechanisms not involving gene silencing. We defined a previously unappreciated activity of miRNAs in inhibiting RNA–protein interactions that, in the context of HIV-1 biology, blocks HIV virus budding and reduces virus infectivity. This occurs by miRNA binding to the nucleocapsid domain of the Gag protein, the main structural component of HIV-1 virions. The resulting miRNA–Gag complexes interfere with viral RNA–mediated Gag assembly and viral budding at the plasma membrane, with imperfectly assembled Gag complexes endocytosed and delivered to lysosomes. The blockade of virus production by miRNA is reversed by adding the miRNA’s target mRNA and stimulated by depleting the endonuclease Argonaute-2 (a component of RISC), suggesting that, when miRNAs are not mediating gene silencing, they can block HIV-1 production through disruption of Gag assembly on membranes. The findings linking miRNA with Gag physiology have significant implications for understanding how cells modulate HIV-1 infection.
Insulin triggers direct trafficking to the cell surface of sequestered GLUT4 storage vesicles marked by Rab10.
Understanding how the glucose transporter isoform 4 (GLUT4) redistributes to the plasma membrane (PM) during insulin stimulation is a major goal of glucose transporter research. GLUT4 molecules normally reside in numerous intracellular compartments, including specialized storage vesicles and early/late endosomes. It is unclear how these diverse compartments respond to insulin stimulation to deliver GLUT4 molecules to the PM. For example, we do not know whether they fuse with each other first or remain as separate compartments with distinct trafficking characteristics. Our recent live-cell imaging studies are helping to clarify these issues. Using Rab proteins (small GTPases involved in the transport of proteins from the Golgi to the PM) as specific markers to distinguish between storage vesicles and endosomes containing GLUT4, we demonstrated that the two pools remain distinct during insulin stimulation and that it is primarily internal GLUT4 storage vesicles (GSVs) marked by Rab10 that approach the plasma membrane and fuse. Our findings add strong support to the model that GSV release from intracellular retention plays a major role in supplying GLUT4 molecules onto the PM under insulin stimulation.
Visualizing cell structure and function with point-localization super-resolution imaging
Fundamental to the success of cell and developmental biology is the ability to tease apart molecular organization in cells and tissues by localizing specific proteins with respect to one another in a native cellular context. However, many key cellular structures (from mitochondrial cristae to nuclear pores) lie below the diffraction limit of visible light, precluding analysis of their organization by conventional approaches. Point-localization super-resolution microscopy techniques such as PALM and stochastic optical reconstruction microscopy (STORM) are poised to resolve, with unprecedented clarity, the organizational principles of macromolecular complexes within cells, thus leading to deeper insights into cellular function in both health and disease. We have been using and improving upon this technology to analyze receptor-protein clustering in the plasma membrane and to characterize subcellular organization. We developed pair-correlation photoactivated localization microscopy (PC-PALM), a technique that uses a pair-correlation algorithm to precisely identify single molecules in super-resolution imaging data sets. The approach enabled us to decipher quantitative features of protein organization within subcellular compartments, including the existence of protein clusters and the size, density, and number of proteins within these clusters. We also employed bleaching/blinking–assisted localization microscopy, which uses conventional fluorophores instead of photoactivatable proteins, to study the nanoscale organization of intracellular structures.
Additional Funding
- NICHD Intramural funding and Pharmacology Research Associate Training program
Publications
- Burnette DT, Shao L, Ott C, Pasapera AM, Fischer RS, Baird MA, Davidson MW, Betzig E, Lippincott-Schwartz J. A contractile and counterbalancing adhesion system controls the 3D shape of crawling cells. J Cell Biol 2014;205:83-96.
- Van Engelenburg S, Shtengel G, Sengupta P, Waki K, Jarnik M, Ablan S, Freed E, Hess H, Lippincott-Schwartz J. Distribution of ESCRT machinery at HIV assembly sites reveals virus scaffolding of ESCRT subunits. Science 2014;343:653-656.
- Satpute-Krishnan P, Ajinkya M, Bhat S, Hegde RS, Lippincott-Schwartz J. ER stress-dependent clearance of misfolded proteins via the secretory pathway. Cell 2014;158:522-533.
- Chen AK, Sengupta P, Waki K, Van Engelenburg SB, Ochiya T, Ablan SD, Freed EO, Lippincott-Schwartz J. MicroRNA binding to HIV-1 Gag protein inhibits Gag assembly and virus production. Proc Natl Acad Sci USA Plus 2014;111:E2676-83.
- Elia N, Ott C, Lippincott-Schwartz J. Incisive imaging and computation for cellular mysteries: lessons from abscission. Cell 2013;155:1220-1231.
Collaborators
- Irwin Arias, MD, Warren Grant Magnuson Clinical Center, NIH, Bethesda, MD
- Eric Betzig, PhD, Howard Hughes Medical Institute, Janelia Farm Research Campus, Ashburn, VA
- Hu Cang, PhD, Waitt Advanced Biophotonics Center, Salk Institute for Biological Studies, La Jolla, CA
- Eric O. Freed, PhD, HIV Drug Resistance Program, Center for Cancer Research, NCI, Frederick, MD
- Dong Fu, PhD, Cell Biology and Metabolism Program, NICHD, Bethesda, MD
- Ramanujan Hegde, MD, PhD, MRC Laboratory of Molecular Biology, Cambridge, United Kingdom
- Harald Hess, PhD, Howard Hughes Medical Institute, Janelia Farm Research Campus, Ashburn, VA
- Peter K. Kim, PhD, The Hospital for Sick Children, Toronto, Canada
- Mark Marsh, PhD, University College London, London, United Kingdom
- Vladislav Verkhusha, PhD, Albert Einstein College of Medicine, New York, NY
- David A. Weitz, PhD, Harvard University School of Engineering and Applied Sciences, Cambridge, MA
Contact
For more information, email lippincj@mail.nih.gov or visit lippincottschwartzlab.nichd.nih.gov.