

You are here: Home > Unit on Microbial Pathogenesis
Virulence Mechanisms of the Human Pathogen Legionella pneumophila

- Matthias Machner, PhD, Head, Unit on Microbial Pathogenesis
- Eric Cheng, PhD, Postdoctoral Fellow
- Byoungkwan Kim, PhD, Postdoctoral Fellow
- Pei-Chung Lee, PhD, Postdoctoral Fellow
- Yi-han Lin, PhD, Postdoctoral Fellow
We investigate the molecular mechanisms by which microbial pathogens exploit the human host and cause disease. As a model system, we use the bacterium Legionella pneumophila, the causative agent of a potentially life-threatening pneumonia called Legionnaires' disease. The disease is primarily found in immune-compromised individuals of any age and acquired after inhalation of contaminated water droplets from L. pneumophila–infested freshwater reservoirs. Once inside the human lung, L. pneumophila is phagocytosed (taken up) by alveolar macrophages. Instead of being degraded by the immune cell, L. pneumophila creates a specialized membrane compartment, the Legionella-containing vacuole (LCV), which supports intracellular bacterial replication (Figure 1). LCV formation depends on the activity of more than 250 proteins, or effectors, which are injected by L. pneumophila into the cytosol of the infected host cell (Figure 1). L. pneumophila mutants that are unable to deliver any effectors fail to escape endolysosomal degradation, underscoring the importance of the effectors' function for bacterial virulence. Our work aims to gain a detailed understanding of the molecular function of the effectors, and how they contribute to L. pneumophila replication within human cells, and will lay the groundwork for the development of novel therapeutic strategies for treating or preventing infections with L. pneumophila and related pathogens.
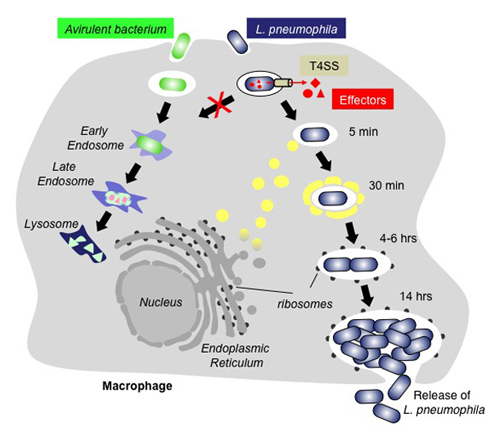
Figure 1. Intracellular replication cycle of L. pneumophila
Whereas avirulent bacteria (green) are rapidly trafficked along the endo-lysosomal route and degraded, L. pneumophila–containing vacuoles (LCV) escape the default endo-lysosomal pathway with the help of the bacterium's effector proteins (red), which are injected into the host cell through the Dot/Icm type IV secretion system (T4SS; beige). Proteins and transport vesicles (yellow spheres) are recruited to the Legionella-containing vacuole (LCV), which is slowly converted into a ribosome-studded compartment that mimics host-cell rough endoplasmic reticulum (ER) and supports replication of L. pneumophila. Eventually, the host cell is lyzed, and L. pneumophila bacteria are released and infect neighboring macrophages.
Regulation of Rab1 through reversible AMPylation
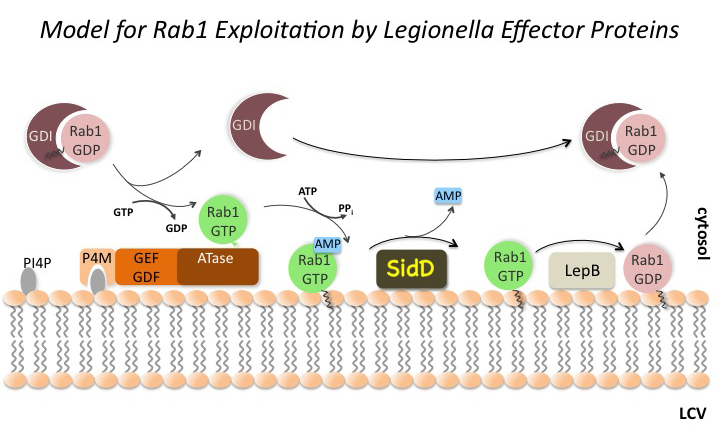
Click image to enlarge.
Figure 2. The Rab1 cycle on LCV membranes
For details see text in section entitled "Regulation of Rab1 through reversible AMPylation."
In order to create a replication vacuole within infected host cells, L. pneumophila has to accomplish a variety of tasks: It must promote the decoration of its LCV with proteins and vesicles from cellular transport routes such as the early secretory pathways regulated by the guanine nucleotide–binding protein (GTPase) Rab1, while at the same time avoiding contact of its LCV with microbicidal endosomes and lysosomes, compartments specialized in cargo degradation. This selective interaction with host-cell compartments is key to infection by L. pneumophila and to obtaining a detailed understanding of the underlying molecular processes is of great scientific interest.
In the past, we studied the molecular mechanisms underlying the recruitment of proteins from the early secretory pathway controlled by the GTPase Rab1 (Figure 2). We and others found that L. pneumophila produces two effectors, SidM and LepB, that mimic eukaryotic GEF and GAP proteins and that activate and later deactivate Rab1 through GDP/GTP exchange and GTP hydrolysis, respectively. Remarkably, our work also demonstrated that the GEF/GAP cycle itself is controlled by a second regulatory circuit, which involves the covalent attachment and removal of adenosine monophosphate (AMP) to Rab1. This process of reversible AMPylation (or adenylylation) is catalyzed by two L. pneumophila effectors with antagonistic activity: the AMPylase SidM and the de-AMPylase SidD. L. pneumophila mutants lacking SidD are unable to catalyze Rab1 de-AMPylation and show a kinetic defect in removing host-cell Rab1 from their LCV. Our study provided the first evidence that microbial pathogens can use post-translational modification in a reversible manner to precisely control the activity of host-cell signaling molecules. Moreover, given that at least some effectors appear to be of eukaryotic origin, as they have been acquired by L. pneumophila from their natural host, freshwater protozoa, it is possible that the regulation of small GTPases through reversible AMPylation is widely distributed among eukaryotes, including humans.
Endosomal avoidance by L. pneumophila
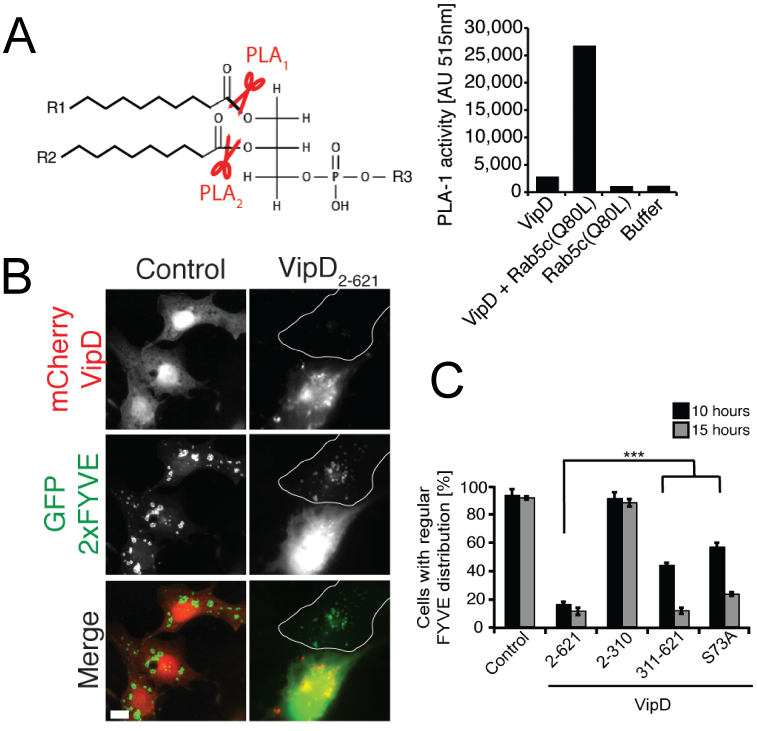
Click image to enlarge.
Figure 3. VipD is a Rab5–activated phospholipase A1 that depletes endosomes of the lipid PI(3)P.
(A) Left: Schematic representation of PLA1 and PLA2 cleavage sites on phospholipids. Right: Fluorometric PLA1 assays using the indicated proteins. The substrate is PED-A1, a BODIPY FL dye–labeled glycerophosphoethanolamine, whose emission is dequenched upon PLA1 hydrolysis. (B) mCherry-VipD, but not the catalytically inactive variant (S73A), displaces the PI(3)P probe GFP-2×FYVE (green) from endosomes. (C) Quantification of the experiment shown in (B). ***P ≤ 0.005 (Student t test).
We also investigated the molecular mechanism underlying endosomal avoidance by the Legionella-containing vacuole (LCV). Phagosomal maturation is a fundamental innate immune mechanism of eukaryotic cells, which facilitates the killing and degradation of ingested microbes. Endosomal fusion is controlled mainly by the GTPase Rab5, which assembles a protein complex that includes the tethering-protein early endosomal antigen 1 (EEA1) and the phosphatidylinositol (PI) 3-kinase hVps34. hVps34 generates PI(3)P, a phospholipid required for membrane association of EEA1 and other endosomal fusion factors. We discovered that the effector protein VipD from L. pneumophila selectively interacts with the active form of Rab5 and Rab22 (another endosomal GTPase). Rab5 binding triggered an otherwise quiescent phospholipase A1 (PLA1) activity in VipD, resulting in the removal of the fatty acid in the sn-1 position of lipids (Figure 3A). When produced within mammalian cells, VipD specifically localized to endosomes, where it catalyzed the removal of PI(3)P from endosomal membranes (Figure 3B,C). EEA1 and other transport and fusion factors that require PI(3)P for membrane binding were consequently depleted from endosomes, most likely rendering the compartment fusion-incompetent. Consequently, we showed that, during host cell infection, VipD significantly reduced exposure of L. pneumophila to the endosomal compartment and protected LCVs from acquiring the endosomal marker Rab5. Our study revealed that, by catalyzing PI(3)P depletion in a Rab5–dependent manner, VipD alters the protein composition of endosomes, thereby blocking their fusion with LCVs.
Structural basis for VipD activation by Rab
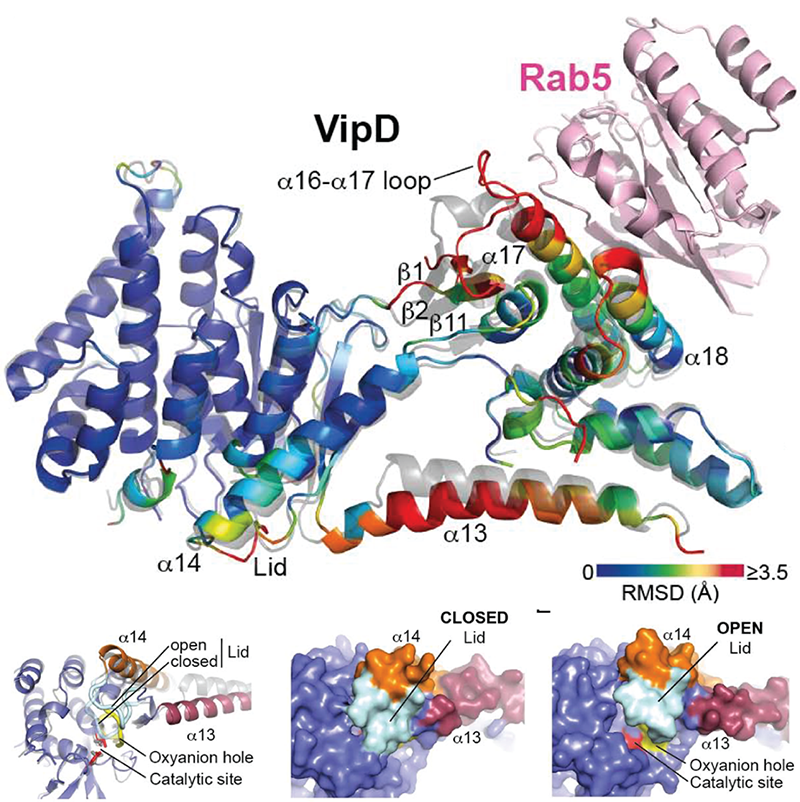
Click image to enlarge.
Figure 4. Allosteric activation of VipD through Rab5 binding.
Top: Structural changes in VipD upon Rab5 binding. Rab518-182 (pink) is complexed to VipD19-564, which is colored from slate to red based on the Root Mean Square Deviation (RMSD) of C-alpha atom pairs when superimposed with the unbound form of VipD19-564 (grey). Below: Close-up view (Left) of the catalytic site, highlighting displacement of the lid (β10-α14 loop, light blue). Surface representation of the unbound (Middle) and Rab5-bound (Right) VipD molecule.
Although T4SS–mediated effector translocation is an efficient and convenient way for pathogens to manipulate host cells from within the safety of their membrane-enclosed compartment, it also creates a challenging dilemma: how the bacteria can ensure that their translocated effectors reach the correct host cell target for manipulation, and how they can prevent effectors from indiscriminately affecting bystander organelles or proteins that may otherwise be beneficial for intracellular survival and replication of the microbe. An emerging theme among microbial effectors is that their enzymatic activity is functionally coupled to their interaction with a particular host factor. To reveal how the phospholipase A1 (PLA1) activity of VipD is triggered upon binding to the host cell GTPase Rab5, we collaborated with the group of Aitor Hierro to determine the crystal structure of VipD in complex with constitutively active Rab5 (Figure 4). We found that an active site–obstructing loop that originates from the C-terminal domain of VipD is repositioned upon Rab5 binding, thereby exposing the catalytic pocket within the N-terminal PLA1 domain. Substitution of individual amino acid residues located within the VipD–Rab5 interface prevented Rab5 binding and PLA1 activation, and caused a failure of VipD mutant proteins to target to Rab5–enriched endosomal structures within cells. The findings disclosed an unexpected mode of long-range allosteric regulation of the PLA1 activity of VipD and provide the basis for the development of novel therapeutic approaches that, rather than directly targeting the enzymes active site, specifically disturb the host factor–mediated activation process of VipD and related microbial phospholipases. The structure of VipD in complex with Rab5c(Q80L) is, to our knowledge, the first to be established of a bacterial phospholipase bound to a host cell protein and the first of any translocated effector in complex with its allosteric activator molecule.
Legionella effectors as biosensors
Several L. pneumophila effectors were shown to contain domains capable of directing them to a particular compartment within the infected host cell. The effector SidM possesses at its C-terminus a domain (P4M) that specifically interacts with the phospholipid PI(4)P. In collaboration with Tamás Balla, we developed an improved version of P4M comprising amino acids 546–647, which was used as a novel biosensor for PI(4)P. We found that the biosensor was capable of detecting pools of the lipid associated not only with the Golgi but also with the plasma membrane and Rab7–positive late endosomes/lysosomes. Therefore, our biosensor P4M shows a wider cellular distribution of PI(4)P than previous probes. The study also highlights the enormous potential of bacterial effectors or their domains as probes for detecting and dissecting the biological function of lipids or other macromolecules within the context of eukaryotic cells.
Additional Funding
- NICHD Director's Challenge Innovation Award
Publications
- Chen Y, Tascón I, Neunuebel MR, Pallara C, Brady J, Kinch LN, Fernández-Recio J, Rojas AL, Machner MP, Hierro A. Structural basis for Rab1 de-AMPylation by the Legionella pneumophila effector SidD. PLoS Pathogens 2013;9:e1003382.
- Neunuebel MR, Mohammadi S, Jarnik M, Machner MP. Legionella pneumophila LidA affects nucleotide binding and activity of the host GTPase Rab1. J Bacteriol 2012;194:1389-1400.
- Gaspar AH, Machner MP. VipD is a Rab5-activated phospholipase A1 that protects Legionella pneumophila from endosomal fusion. Proc Natl Acad Sci USA 2014;111:4560-4565.
- Hammond GR, Machner MP, Balla T. A novel probe for phosphatidylinositol 4-phosphate reveals multiple pools beyond the Golgi. J Cell Biol 2014;205:113-126.
- Lucas M, Gaspar AH, Pallara C, Rojas AL, Fernández-Recio J, Machner MP, Hierro A. Structural basis for the recruitment and activation of the Legionella phospholipase VipD by the host GTPase Rab5. Proc Natl Acad Sci USA 2014;111:E3514-3523.
Collaborators
- Tamás Balla, PhD, Program in Developmental Neuroscience, NICHD, Bethesda, MD
- Howard Hang, PhD, The Rockefeller University, New York, NY
- Aitor Hierro, PhD, CIC bioGUNE Institute, Bilbao, Spain
- Joshua LaBaer, MD, PhD, Virginia G. Piper Center for Personalized Diagnostics, Arizona State University, Tempe, AZ
Contact
For more information, email machnerm@mail.nih.gov or visit cbmp.nichd.nih.gov/ump/research.html