
Structural and Chemical Biology

- Anirban Banerjee,
PhD, Head, Section on Structural and Chemical Biology - Mitra Rana, PhD, Staff Scientist
- Hyojung Kim, PhD, Visiting Fellow
- Chandrashekhar Kumar, PhD, Visiting Fellow
- Tanmay Mondal, PhD, Visiting Fellow
- Rahul Raina, PhD, Visiting Fellow
- Ateek Shah, PhD, Visiting Fellow
- Robert Hall, PhD, Postdoctoral Intramural Research Training Award Fellow
- Christian Knott, BS, Postbaccalaureate Fellow
For all living systems, it is essential for their internal environment, where the processes that sustain and propagate life occur, to remain separated from the outside environment. This separation is achieved by the cell membrane, acting as a barrier and thereby creating distinct cellular compartments for biological processes (Figure 1). Cell membranes are primarily composed of lipids, chemical components that do not mix with the water-based interior of the cell, just as oil and water do not mix. However, the cell membrane is far from being a passive barrier. It is very much an active part of cell-based life, containing resident proteins that carry out a wide range of functions. Proteins are the workhorses of cells and carry out the majority of the functions in living organisms. Proteins that reside within membranes catalyze chemical transformations, communicate between the two sides of the membrane, and move cargo across the membrane. They account for about 25% of all proteins but constitute more than 60% of drug targets. They are also involved in devastating diseases, such as cancer, cardiovascular diseases, and neurodegenerative disorders, to name just a few. Despite their outsized importance in human health and disease, precise details of how many membrane proteins perform their functions are poorly understood. This is because they are challenging to study, starting from obtaining enough material for conducting experiments. We are interested in two broadly different groups of membrane proteins: the first attach lipids to other proteins, and the second move small charged ions and molecules across the membrane. Although they have been linked to diseases such as Huntington’s disease, pancreatic cancer and Friedreich’s ataxia, the detailed understanding of how they work remains unclear. We are employing a range of tools, including high-resolution structure solution, chemical biology, cell biology, biochemistry, biophysics, and proteomics to understand how they function. We aspire to reveal insights into how their malfunctioning contributes to human disease and to pave the way for new therapies that can promote human health and well-being.
Figure 1. Simple depiction of a eukaryotic cell, the cell membrane, and few representative membrane-embedded proteins
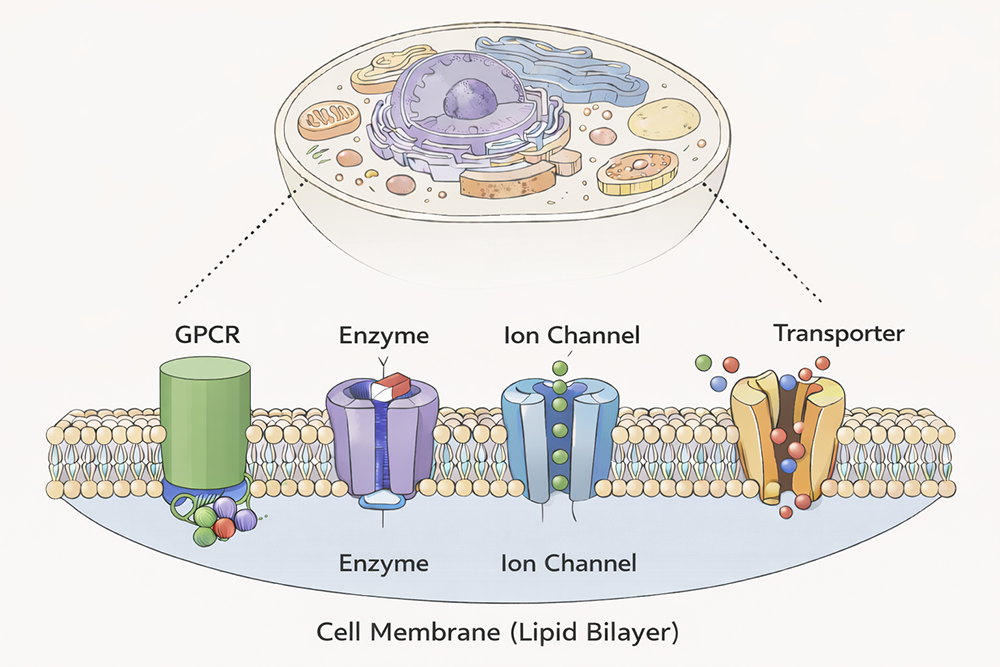
Figure 1. Simple depiction of a eukaryotic cell, the cell membrane, and few representative membrane-embedded proteins
Molecular underpinnings of protein palmitoylation (protein S-acylation): the most common form of protein lipidation
Co- and post-translational modifications greatly expand the structural, chemical, and functional diversity of the proteome. Of these, protein lipidation, which collectively refers to covalent modification of proteins by lipids, constitutes a centrally important class of post-translational modifications. Protein S-acylation, commonly known as protein palmitoylation, is a specific form of protein lipidation whereby long-chain fatty acids, typically C16 palmitic acid, become covalently attached to cytosol-facing cysteines through a thioester linkage. S-acylation is one of the most pervasive and physiologically important post-translational modifications, with thousands of substrates ranging from ion channels to cell-surface receptors, neuronal scaffolding proteins, and small GTPases.
The physico-chemical effect of S-acylation is to alter the local hydrophobicity of the substrate protein. The thioester bond makes S-acylation unique in being a relatively labile moiety, and it can be cleaved by protein S-thioesterase enzymes. This makes S-acylation the only reversible form of protein lipidation and one of the few dynamic post-translational modifications. The physiological effects of S-acylation are diverse and are of critical cellular importance. For example, Ras, a small GTPase that is critical for cellular growth and differentiation and is mutated in about one-third of all human cancers, is S-acylated at the Golgi apparatus and subsequently targeted to the plasma membrane by vesicular transport. S-acylated Ras localizes to cholesterol-rich domains on the plasma membrane. However, it is subsequently deacylated by the thioesterase APT1, dissociates from the plasma membrane, and redistributes on endomembranes, including the Golgi. Such dynamic recycling of Ras is critical for its function. In recent work, we also showed that the Spike protein of SARS-CoV-2, the causative agent of COVID-19, is S-acylated, which is important in the viral life cycle.
Protein S-acylation is catalyzed by a large group of enzymes known as zDHHC palmitoyl acyltransferases (also referred to as DHHC enzymes or DHHC-PAT), so named because they contain a signature D-H-H-C motif (aspartate-histidine-histidine-cysteine) in a cysteine-rich domain (CRD) in an intracellular loop (Figure 2). These are polytopic, integral membrane proteins localized to the membranes of a variety of cellular compartments. Humans have 23 zDHHC proteins encoded in their genome. Beyond the shared DHHC domain, zDHHC proteins vary considerably; some possess ankyrin repeats (structural protein motifs that mediate protein-protein interactions), a few have six transmembrane helices instead of the usual four, and some form functionally relevant heterodimers with cytoplasmic auxiliary subunits.
Figure 2. Organization and properties of DHHC palmitoyltransferases (PATs)
a. The organization of three different DHHC-PATs is shown schematically. The spheres indicate protein-protein interaction domains. Erf2 associates with a cytoplasmic subunit, Erf4, to form the active enzyme. b. The DHHC-CRD (cysteine-rich domain) region of a few representative DHHC-PATs are aligned. The conserved amino acids are shown in red. c. Reaction catalyzed by DHHC-PATs; the reverse reaction is catalyzed by acylprotein thioesterases (APT).
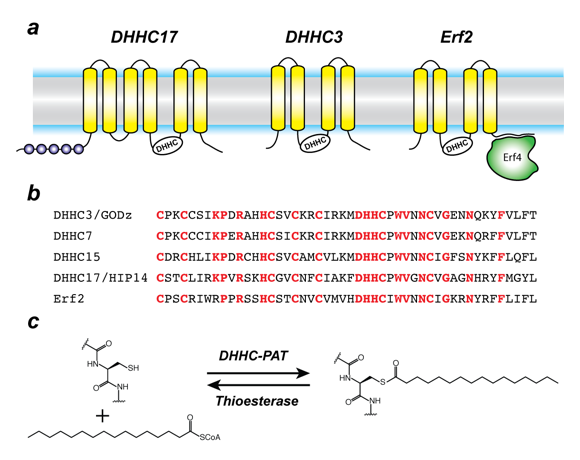
Figure 2. Organization and properties of DHHC palmitoyltransferases (PATs)
a. The organization of three different DHHC-PATs is shown schematically. The spheres indicate protein-protein interaction domains. Erf2 associates with a cytoplasmic subunit, Erf4, to form the active enzyme. b. The DHHC-CRD (cysteine-rich domain) region of a few representative DHHC-PATs are aligned. The conserved amino acids are shown in red. c. Reaction catalyzed by DHHC-PATs; the reverse reaction is catalyzed by acylprotein thioesterases (APT).
Thousands of substrates of zDHHC enzymes have been identified. Yet, to date, no consensus sequence has been identified for protein S-acylation. A specific zDHHC enzyme can S-acylate many substrates, and, conversely, a given substrate can be S-acylated by many zDHHC enzymes. Such redundancy has been one of the most intriguing aspects of protein S-acylation and makes it difficult to assign substrates by overexpression/knockout strategies, given that, in the absence of one zDHHC enzyme, other zDHHC family members can take over. However, this does not necessarily reflect the true enzyme-substrate relationship. The problem is even more confounded by the lack of specific inhibitors of zDHHC enzymes. Even though 2-bromopalmitate is widely used as a global inhibitor of zDHHC enzymes, it has been shown that it also broadly targets other proteins involved in lipid metabolism.
Besides its broad importance in cell biology, S-acylation has been increasingly connected to many devastating human diseases. Mutations in zDHHC9 are implicated in X-linked intellectual disability. EGFR (epidermal growth factor receptor) localization is regulated by S-acylation by zDHHC20, which plays plays a critical role in signal regulation during oncogenesis. This makes zDHHC20 a target for developing new therapies against a variety of cancers. It was shown that zDHHC20 S-acylates EGFR and is thus a potential therapeutic target in a variety of cancers. The protein huntingtin (HTT) is S-acylated by zDHHC17 (and zDHHC13). Reduced S-acylation in mutant HTT is linked to the neurodegenerative disorder Huntington disease. Palmitoylprotein thioesterase-1 (PPT1) loss-of-function causes infantile neuronal ceroid lipofuscinosis (CLN1/INCL), a severe childhood neurodegenerative disorder. Thus, there are tantalizing connections between protein S-acylation and neurodegeneration and neuroinflammation, to name a few examples. However, despite their importance across a broad spectrum of biological pathways and their biomedical importance, nothing was known about the structural mechanism of zDHHC S-acyltransferases when we started working on this family.
We began working on the structures and mechanisms of zDHHC enzymes. In 2018, we published the first high-resolution structures of two members of the zDHHC family: human zDHHC20 and the zebrafish homolog of zDHHC15. An outstanding question in the field of protein S-acylation is the nature of zDHHC–substrate interactions, which has remained poorly understood for most zDHHC enzymes. Cell-based experiments indicate a promiscuous and complex zDHHC–substrate network, whereas lack of in vitro reconstitution experiments has impeded insights into the nature of discrete zDHHC–substrate interactions. In vitro substrate S-acylation assays can also be developed for high-throughput assays for discovery of inhibitors of zDHHC enzymes, which can be the starting point for drug discovery projects. With this in mind, we recently began focusing on developing in vitro substrate S-acylation assays of zDHHC enzymes.
We designed a novel substrate S-acylation reconstitution assay called the Pep-PAT assay, using purified enzyme and peptide fragments of substrates (Figure 3). We used the Pep-PAT assay to investigate the substrate S-acylation of three different zDHHC enzymes on seven different substrates. Remarkably, all the zDHHC enzymes showed robust activity with certain substrates but not others (Figure 3). These in vitro reconstitution experiments indicate that there is a preferred substrate hierarchy for zDHHC enzymes. We further used the Pep-PAT assay to interrogate the role of neighboring residues around the target cysteine on S-acylation of PSD-95 (postsynaptic density protein 95)and SARS-CoV-2 spike protein. These experiments revealed that select residues around the target cysteines have a distinct impact on substrate S-acylation, leading to the first insights into how neighboring residues around the target cysteine affect substrate S-acylation by zDHHC enzymes. This was the first report of reconstituting substrate selectivity of zDHHC enzymes in vitro, and builds a framework for understanding the interactions between zDHHC enzymes and their substrates.
Figure 3. Pep-PAT assay for in vitro reconstitution of the substrate selectivity of zDHHC enzymes
a. A substrate S-acylation assay, which we call the Pep-PAT assay, using purified zDHHC enzymes and synthetic peptide fragments of substrates and coupling the released CoASH (coenzyme A) with a second enzyme to generate stoichiometric amount of NADH, which is fluorescent. b. The design of the substrate fragments involve attachment of an N-terminal myristoyl moiety to mimic the local hydrophobic environment of the target cysteine. c. Pep-PAT assay showing that human zDHHC20 has robust activity on SARS-CoV-2 spike protein but not PSD-95 (postsynaptic density protein 95). d. Pep-PAT assay showing that human zDHHC15 has robust activity on the same PSD-95 substrate as in c.
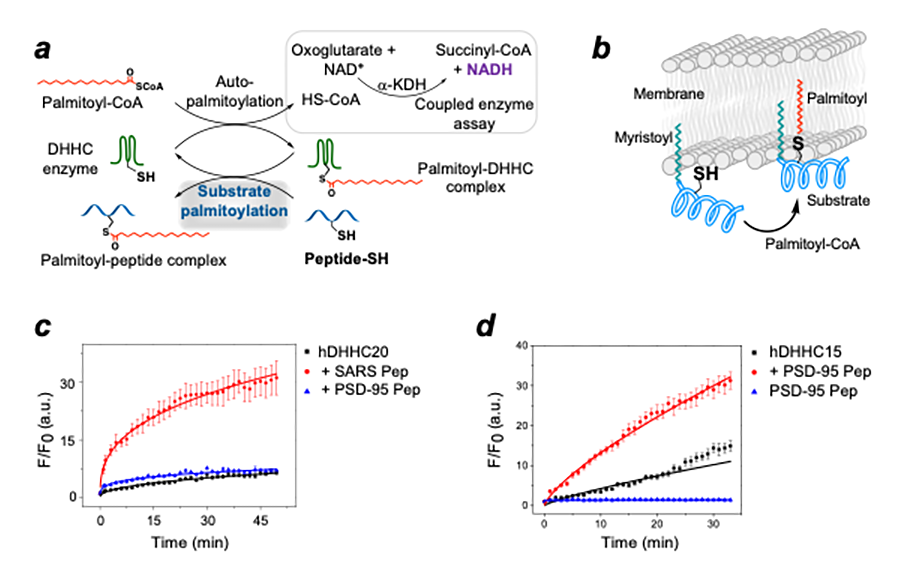
Figure 3. Pep-PAT assay for in vitro reconstitution of the substrate selectivity of zDHHC enzymes
a. A substrate S-acylation assay, which we call the Pep-PAT assay, using purified zDHHC enzymes and synthetic peptide fragments of substrates and coupling the released CoASH (coenzyme A) with a second enzyme to generate stoichiometric amount of NADH, which is fluorescent. b. The design of the substrate fragments involve attachment of an N-terminal myristoyl moiety to mimic the local hydrophobic environment of the target cysteine. c. Pep-PAT assay showing that human zDHHC20 has robust activity on SARS-CoV-2 spike protein but not PSD-95 (postsynaptic density protein 95). d. Pep-PAT assay showing that human zDHHC15 has robust activity on the same PSD-95 substrate as in c.
The Pep-PAT assay is a coupled enzyme assay and thus an indirect readout of zDHHC activity. More recently, we reconstituted zDHHC activity in membrane environments using lipid nanodiscs in a direct fluorescence polarization-based assay, using fluorescently labelled peptide substrates and purified enzymes. We used this assay to demonstrate the role of cholesterol in modulating zDHHC activity. We also designed a Giant Unilamellar Vesicle (GUV)–based assay, which permits a direct visualization-based measurement of zDHHC activity. The biochemical functions of most members of the zDHHC family are poorly characterized, even though there are emerging connections to human diseases. Our assay platforms will permit us to unravel the structural and molecular mechanism of these uncharacterized zDHHC enzymes, and will also open up avenues for discovering selective small-molecule inhibitors that would be powerful cell-biological tools as well as possible starting points for drug discovery.
Another major challenge in the area of protein S-acylation has been identification of S-acylated proteins. There are no sequence specifiers of S-acylation except that membrane-proximal cytosol-facing cysteines have a higher propensity to be S-acylated. However, membrane proximal protein segments are often hydrophobic and are not identified in proteomic analyses. In collaboration with Aleksandra Nita-Lazar, we have been investigating efficient identification of S-acylated proteins. In a recent manuscript, we introduced DDM, a mild detergent and the workhorse in membrane-protein structural biology, in the workflow to identify S-acylated proteins. In HEK-293 cells (an immortalized cell line), we demonstrated that the technique led to identification of hundreds of new S-acylated proteins (Figure 4).
Figure 4. Workflow for enhanced discovery of palmitoylated proteins
Cultured cells lysed in 2% SDS buffer were processed through modified acyl biotin exchange (ABE) steps: first, free cysteines were blocked by N-ethylmaleimide (NEM), then palmitoylated cysteines were cleaved by hydroxylamine, followed by labeling with biotin-HPDP (a sulfhydryl-reactive biotinylation agent) of the resultant free cysteines from hydroxylamine cleavage. After streptavidin enrichment and TCEP elution, chloroform-methanol precipitation was performed. Precipitated proteins were resolubilized with either 8 M urea alone or 8 M urea supplemented with 0.05% or 0.1% DDM before reduction, alkylation, and tryptic digestion for LC-MS/MS (liquid chromatography–tandem mass spectrometry) analysis. To enhance discovery of palmitoylated proteins, in an alternate workflow, Asp-N, chymotrypsin, or Glu-C was used.
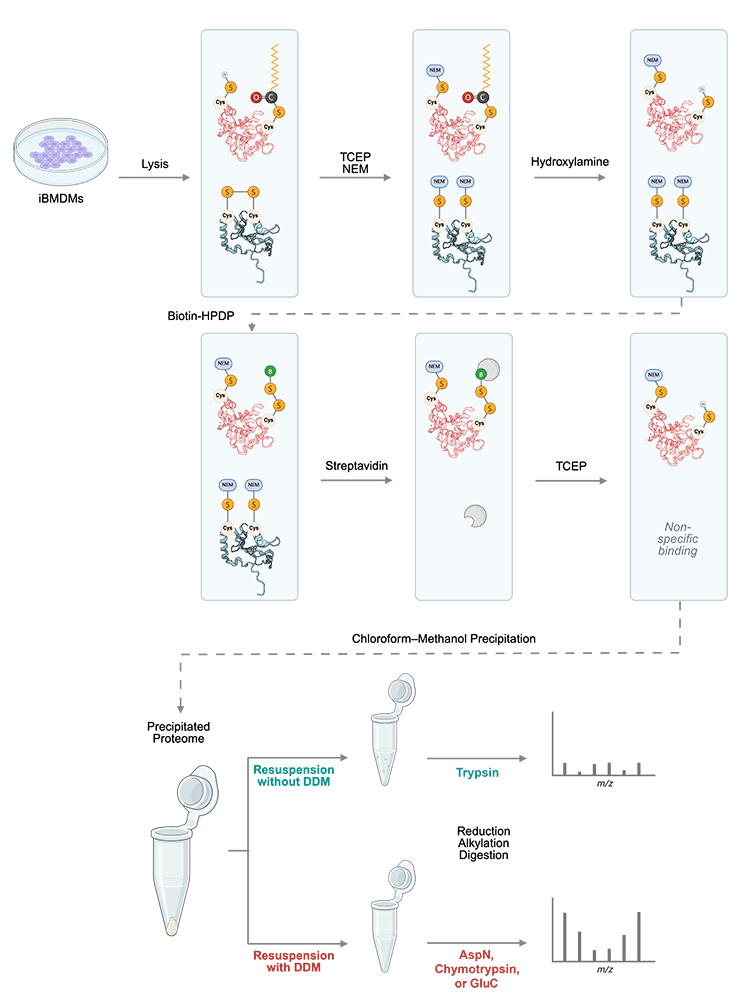
Figure 4. Workflow for enhanced discovery of palmitoylated proteins
Cultured cells lysed in 2% SDS buffer were processed through modified acyl biotin exchange (ABE) steps: first, free cysteines were blocked by N-ethylmaleimide (NEM), then palmitoylated cysteines were cleaved by hydroxylamine, followed by labeling with biotin-HPDP (a sulfhydryl-reactive biotinylation agent) of the resultant free cysteines from hydroxylamine cleavage. After streptavidin enrichment and TCEP elution, chloroform-methanol precipitation was performed. Precipitated proteins were resolubilized with either 8 M urea alone or 8 M urea supplemented with 0.05% or 0.1% DDM before reduction, alkylation, and tryptic digestion for LC-MS/MS (liquid chromatography–tandem mass spectrometry) analysis. To enhance discovery of palmitoylated proteins, in an alternate workflow, Asp-N, chymotrypsin, or Glu-C was used.
In another related manuscript [Reference 2], we reasoned that the trypsin cleavage conditions, used for generating peptides in the standard proteomic workflow, rely on the presence of Lys and Arg residues, which may not be present in the hydrophobic environment around all cysteines that are S-acylated. We employed a comprehensive multi-protease strategy (using trypsin, the endoproteinase AspN, chymotrypsin, and the serine protease GluC) coupled with acyl-biotin exchange (ABE) (Figure 4). We also moved beyond the commonly used RAW 264.7 cells, employing immortalized bone marrow-derived macrophages (iBMDMs) to generate the first deep palmitoylome map in a model that closely mimics primary macrophage physiology. We identified over 2,500 putative S-palmitoylated proteins in immune cells, including over 1,000 candidates not previously reported in mouse immune cells. Crucially, a significant number of these were identified exclusively by the non-tryptic proteases. The study also revealed how rapidly the palmitoylome remodels within just 30 minutes of immune activation by the lipopolysaccharide LPS.
Molecular mechanism of iron transport across cellular membranes
Transition metals such as iron and copper perform essential functions across all kingdoms of life. Their versatile coordination environment as well as the ability to take part in redox processes make them particularly important in a very wide range of physiological processes. We are interested in how transition metal ions are transported across the membrane. Currently, we are focusing on iron transporters because of the importance of iron in human health and disease. Iron is a key part of heme, the cofactor that enables oxygen transport by hemoglobin, and is an essential cofactor for many other important enzymes. Iron-containing cofactors called iron-sulfur clusters are at the heart of electron transfer during respiration. Dysregulation in iron transport and homeostasis is linked to many devastating diseases. Iron-deficiency anemia is associated with neurodevelopmental delays, impaired cognitive and motor function, and poor growth in infants and children. Mutations of the iron transporter DMT1 are linked to microcytic anemia with iron overload. Iron is also a major axis of interaction between pathogens and host, given that many pathogens rely on iron for survival and proliferation, and that several aspects of human iron metabolism ensure that iron is scarcely accessible to pathogens.
In higher organisms, mitochondria are the ‘hotspot’ for the cell biology of iron, because, there, Fe-S clusters are biosynthesized and iron is inserted into heme. Mitochondrial iron homeostasis plays a critical role in cellular iron homeostasis and in the overall physiology of the cell. In vertebrates, the only known major transporters of iron into mitochondria are mitoferrin-1 and mitoferrin-2, two homologous members of a large group of mitochondrial transporters known as the mitochondrial carrier family (Figure 5a). Mitoferrin-1 (Mfrn1) is expressed mainly in erythroid cells, while mitoferrin-2 (Mfrn2) is expressed ubiquitously. Knockout of Mfrn1 is embryonically lethal, reflecting the importance of mitoferrins in vertebrate physiology.
Figure 5. Transporters of iron
a. Iron is imported through the plasma membrane by the transferrin/transferrin receptor (blue) cycle and is transported out of endosomes by the divalent metal ion transporter (DMT) (grey); iron is delivered to mitoferrin (yellow cylinders) by unknown means; mitoferrin delivers iron to unknown partners in mitochondria, which become available for heme and Fe-S cluster biosynthesis. b. Schematic showing Legionella entering a host cell and sequestering itself in a Legionella-containing vacuole (LCV): MavN is inserted into the membrane of the LCV and hijacks iron from the host cell.
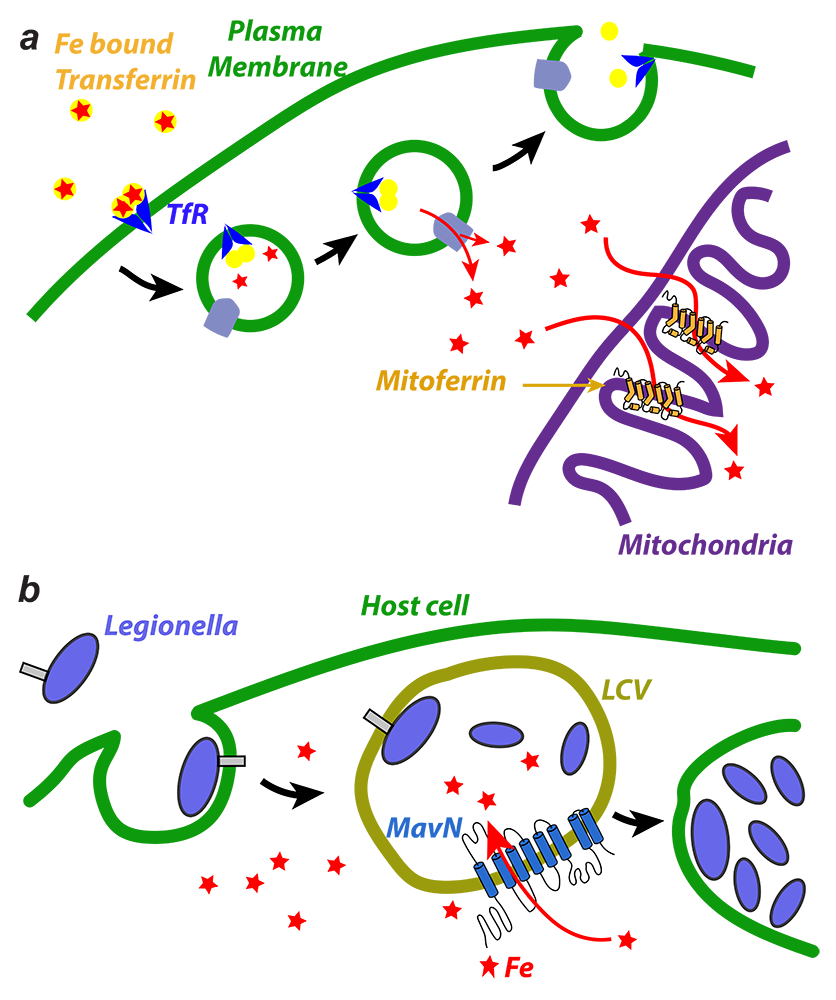
Figure 5. Transporters of iron
a. Iron is imported through the plasma membrane by the transferrin/transferrin receptor (blue) cycle and is transported out of endosomes by the divalent metal ion transporter (DMT) (grey); iron is delivered to mitoferrin (yellow cylinders) by unknown means; mitoferrin delivers iron to unknown partners in mitochondria, which become available for heme and Fe-S cluster biosynthesis. b. Schematic showing Legionella entering a host cell and sequestering itself in a Legionella-containing vacuole (LCV): MavN is inserted into the membrane of the LCV and hijacks iron from the host cell.
Mfrn1 and Mfrn2 were discovered more than 15 years ago. However, the proposed iron-transport activity had not been demonstrated using an in vitro functional reconstitution assay, and nothing was known about their interaction with iron or other related metal ions, most likely because heterologous overexpression and purification of mitoferrins were not reported in the literature. There were also no reports of a reconstituted iron-transport assay starting from purified protein. In earlier work, we carried out heterologous purification, in vitro functional reconstitution, and mutational dissection of a vertebrate Mfrn1, the first demonstration that Mfrn1 can indeed transport iron. Our studies provided the first biochemical insights into Mfrn function. In earlier work, we also used our in vitro proteoliposome-reconstituted iron-transport assay, the first such assay to be reported in the literature, to dissect the iron-transport activity of MavN, another highly conserved iron transporter, in the bacterial pathogen Legionella pneumophila (Figure 5b). Legionella pneumophila is the causative agent of Legionnaire's disease, a form of pneumonia that particularly affects older and immune-compromised individuals. We are currently working towards understanding the detailed mechanism of iron recognition and transport by Mfrn1 and MavN.
Additional Funding
- Office of Dietary Supplements
Publications
- In vitro reconstitution reveals substrate selectivity of protein S-acyltransferases. J Biol Chem 2025 301(4):108406
- Proteome-wide analysis of palmitoylated proteins in macrophages reveals novel insights into early immune signaling. Proteomics 2025 e70100; online ahead of print
- Enhanced S-palmitoylated protein detection by mild nonionic detergent in proteomic workflow. J Am Soc Mass Spectrom 2025 37(1):64-73
Collaborators
- Angela Ballesteros, PhD, Section on Sensory Physiology and Biophysics, NIDCD, Bethesda, MD
- Thomas Dever, PhD, Division of Molecular and Cellular Biology, NICHD, Bethesda, MD
- Rodolfo Ghirlando, PhD, Laboratory of Molecular Biology, NIDDK, Bethesda, MD
- Kallol Gupta, PhD, Yale University, New Haven, CT
- James Inglese, PhD, Preclinical Chemical Biology Laboratory, NCATS, Rockville, MD
- Ralph Isberg, PhD, Tufts University School of Medicine, Boston, MA
- Claire Le Pichon, PhD, Neurosciences and Cellular and Structural Biology Division, NICHD, Bethesda, MD
- Yan Li, PhD, Core Director, Protein, Peptide Sequencing Facility, NINDS, Bethesda, MD
- Aleksandra Nita-Lazar, PhD, Laboratory of Immune System Biology, NIAID, Bethesda, MD
- Meru Sadhu, PhD, Genetic Disease Research Branch, NHGRI, Bethesda, MD
- Lei Shi, PhD, Molecular Targets and Medications Discovery Branch, NIDA, Baltimore, MD
- Rolf Swenson, PhD, The Chemistry and Synthesis Center, NHLBI, Rockville, MD
- Yihong Ye, PhD, Laboratory of Molecular Biology, NIDDK, Bethesda, MD
Contact
For more information, email anirban.banerjee@nih.gov or visit https://www.nichd.nih.gov/research/atNICHD/Investigators/banerjee.