
Regulation of Childhood Growth

- Jeffrey Baron,
MD, Chief, Section on Growth and Development - Lesley Brown, PhD, Staff Scientist
- Julian Lui, PhD, Staff Scientist
- Krishma Tailor, PhD, Biologist
- Wei Wang, MSc, Biologist
- Kirtal Hansdah, PhD, Visiting Fellow
- Isabelle Hannula, BS, Postbaccalaureate Fellow
- Frema Owusu-Ansah, BS, Postbaccalaureate Fellow
- Arun Rama-Krishnan, BS, Postbaccalaureate Fellow
- Aidan Remy, BS, Postbaccalaureate Fellow
We investigate the cellular and molecular mechanisms governing childhood growth. We focus particularly on skeletal growth at the growth plate, a thin layer of cartilage found near the ends of juvenile bones (Figure 1). In the growth plates, chondrocyte proliferation, hypertrophy, and cartilage matrix synthesis result in chondrogenesis, the production of new cartilage. The newly formed cartilage is remodeled into bone. The net result is that new bone is progressively created adjacent to the growth plates, which causes bones to elongate and therefore causes children to grow in height.
Figure 1. Histological image of a growth plate, showing the three principal zones
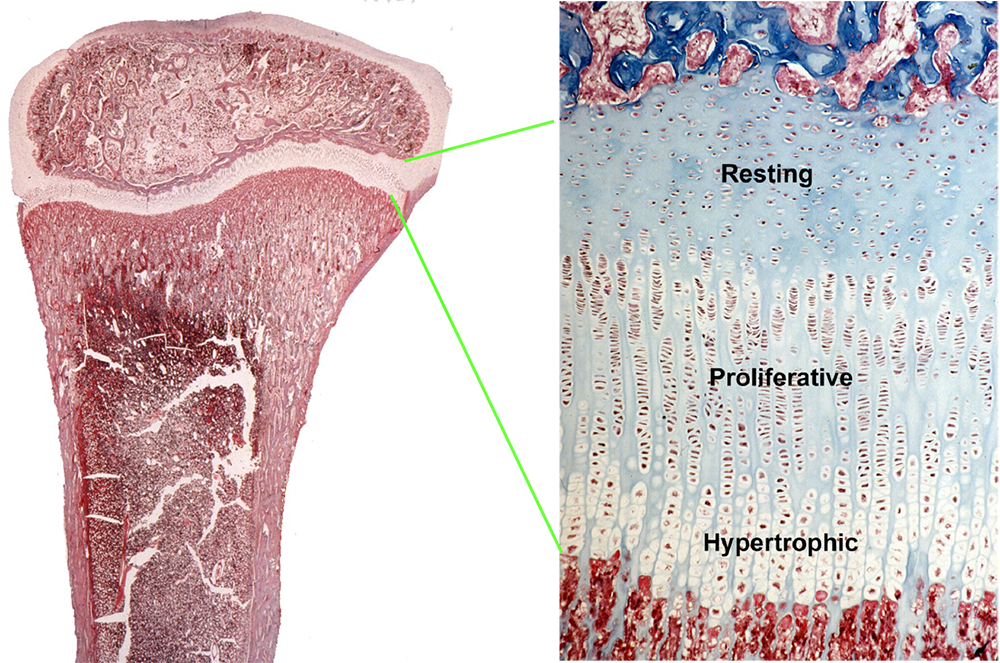
Figure 1. Histological image of a growth plate, showing the three principal zones
One goal of this work is to gain insight into the many human genetic disorders that cause childhood growth failure or overgrowth. A second goal is to develop new treatments for children with severe growth disorders.
Novel genetic causes of childhood growth disorders
Genetic variants that impair growth-plate chondrogenesis cause short stature, whereas variants that increase growth-plate chondrogenesis cause tall stature. In some of these disorders, the bones are malformed, in which case the condition is termed a skeletal dysplasia. Some genetic disorders affect other tissues in addition to the growth-plate cartilage, producing a more complex genetic syndrome.
For many children with growth disorders, clinical, laboratory, and genetic evaluation fails to identify the underlying etiology. To discover new genetic causes of skeletal growth disorders, we studied families with inherited conditions affecting growth, using exome sequencing to detect causative genetic variants. The identified sequence variants and the genes in which they occur were studied in the laboratory to confirm that the variant is pathogenic, to elucidate the pathogenesis of the disorder, and to explore the role of the gene in normal growth.
Using this approach, our group identified several new causes of disorders that impair skeletal growth. We found that variants in the QRICH1 gene (encoding glutamine-rich protein 1, a transcriptional regulator) cause a chondrodysplasia attributable to impaired growth-plate chondrocyte hypertrophic differentiation. We also discovered that variants in aggrecan (encoded by ACAN), a component of cartilage extracellular matrix, cause autosomal-dominant short stature with advanced skeletal maturation and that these patients tend to develop early-onset osteoarthritis. We also found that a neomorphic mutation in SP7, which encodes a transcription factor, causes a complex skeletal dysplasia that includes severe scoliosis, thickened calvarium, craniosynostosis, osteosclerosis, and bone fragility [Reference 1]. The variant altered the DNA–binding sequence specificity of the transcription factor, causing aberrant expression of osteoblast-related genes, abnormal osteoblast differentiation, and abnormal bone formation [Reference 1]. Recently, we discovered that variants in WASHC3 (WASHC3 is a component of the WASH complex, which is involved in endosome receptor recycling) cause short stature, neurodevelopmental abnormalities, and distinctive facial appearances [Reference 2]. We found evidence that the variants affected endosomal trafficking, signal transduction by the PTH1R receptor (a receptor for parathyroid hormone), and hypertrophic differentiation of growth-plate chondrocytes [Reference 2].
We also investigated disorders that cause excessive skeletal growth. One such overgrowth disorder, Weaver syndrome, is caused by variants in EZH2, which encodes a histone methyltransferase and thereby acts as an epigenetic writer. We found that the EZH2 variants responsible for Weaver syndrome cause a partial loss of enzymatic function [Reference 3].
We discovered that loss-of-function variants in Spindlin 4 (SPIN4) cause a generalized overgrowth syndrome of prenatal onset with an X-linked inheritance. Ablation of Spin4 in mice recapitulated the human phenotype. We found evidence that SPIN4 binds to specific histone modifications, promotes canonical WNT signaling, inhibits cell proliferation in the growth plate, and negatively regulates the number of progenitor chondrocytes in the resting zone (Figure 2). Taken together, our findings provide strong evidence that SPIN4 is an epigenetic reader that negatively regulates mammalian body growth, and that loss of SPIN4 causes an overgrowth syndrome in humans [Reference 4].
Figure 2. Diagram depicting the effects of Spin4 on skeletal growth
Spin4 binds to specific histone modifications, promotes canonical WNT signaling, inhibits cell proliferation in the growth plate, and negatively regulates the number of progenitor chondrocytes in the resting zone.
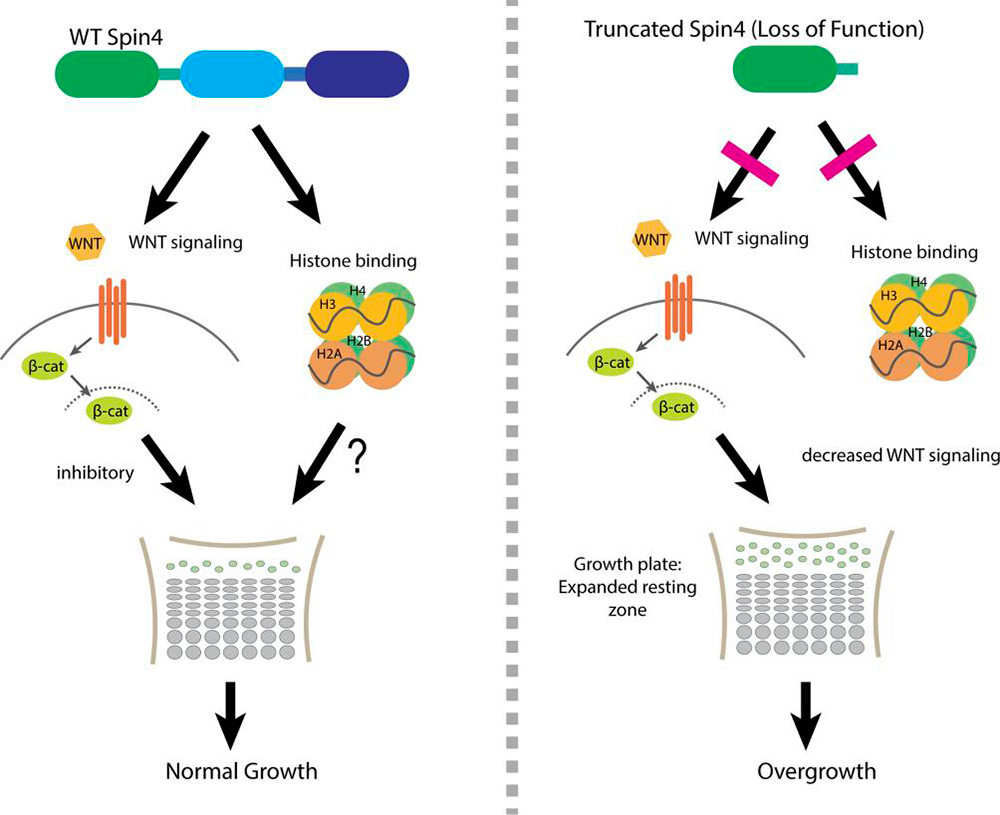
Figure 2. Diagram depicting the effects of Spin4 on skeletal growth
Spin4 binds to specific histone modifications, promotes canonical WNT signaling, inhibits cell proliferation in the growth plate, and negatively regulates the number of progenitor chondrocytes in the resting zone.
We continued to explore the phenotype of this condition in a mouse model. Because many overgrowth syndromes are associated with increased risk of tumorigenesis, we investigated the prevalence of neoplasms, along with body composition and bone mineral density, in aged Spin4 knockout mice. We found that male mice lacking Spin4 have an increased number of tumors, while body composition and bone mineral density were comparable to wild-type mice. We also analyzed publicly available expression data and found that the expression of SPIN4 was elevated in many human cancers compared with the corresponding non-malignant tissue samples. Our findings suggest that loss of SPIN4 may predispose to neoplasia but does not appear to affect adiposity or bone density.
Molecular and cellular mechanisms by which specific genes and pathways regulate childhood growth
Our group also studies the fundamental mechanisms governing childhood growth. Much of our work has focused on the growth plate. Growth plate chondrogenesis is controlled by many endocrine, paracrine, extracellular matrix–related, and intracellular regulatory systems. Our group has helped elucidate growth-plate regulation by FGFs (fibroblast growth factors), BMPs (bone morphogenetic proteins), C-type natriuretic peptide, retinoids, WNT signaling pathways, the PTHrP/IHH feedback loop, IGFs (insulin-like growth factors), estrogens, glucocorticoids, transcription factors such as SOX9, microRNAs, and epigenetic mechanisms.
Recently, we continued to explore the molecular and cellular mechanisms by which Spin4 regulates skeletal growth. We found evidence that postnatal loss of Spin4 deletion in chondroprogenitor cells in the growth plate is sufficient to increase bone growth. We also found evidence that the effect of Spin4 on growth is mediated by interaction between Spin4 and specific modified histones, indicating that Spin4 acts as an epigenetic reader to control body size, as indicated above.
New treatment approaches for growth-plate disorders
Recombinant human growth hormone (GH) is commonly used to treat short stature in children. However, GH treatment has limited efficacy, particularly in severe, non-GH–deficient conditions such as chondrodysplasias, and has off-target effects. Systemic insulin-like growth factor-1 (IGF-1) treatment has similar deficiencies. There are many endocrine and paracrine factors that promote chondrogenesis at the growth plate that could potentially be used to treat these disorders. Targeting these growth factors specifically to the growth plate might augment the therapeutic skeletal effect while diminishing undesirable effects on non-target tissues. To develop growth plate–targeted therapy, we previously used yeast display to identify single-chain human antibody fragments that bind to cartilage with high affinity and specificity. We then created fusion proteins combining these cartilage-targeting antibody fragments with IGF-1, an endocrine/paracrine factor that positively regulates chondrogenesis. Using a GH–deficient mouse model, we found that subcutaneous injections of these fusion proteins increased growth-plate height without increasing proliferation in kidney cortical cells, demonstrating greater on-target efficacy at the growth plate and less off-target effect on the kidney than IGF-1 alone. Our findings provide proof of principle that targeting therapeutics to growth plate cartilage can potentially improve treatment for childhood growth disorders.
These fusion proteins might be particularly well suited to treat GH insensitivity (Laron) syndrome. In this disorder, the current treatment with rIGF-1 has incomplete efficacy, substantial adverse effects including hypoglycemia, and requires twice-daily injections. We therefore studied the effects of cartilage-targeted IGF-1 in a mouse model of GH resistance (achieved with pegvisomant) [Reference 5]. We showed that our IGF-1–ctAb fusion protein had efficacy at the growth plate similar to that seen in the GH–deficient mouse model. A once-daily injection of IGF-1–ctAb showed efficacy at least as great as twice-daily IGF-1. We also found that subcutaneous IGF-1–ctAb had a lower hypoglycemic effect than IGF-1. In vitro, IGF-1–ctAb caused a prolonged increase in IGF receptor (pAKT) signaling compared with IGF-1, and this effect was dependent on matrilin-3 (the specific targeted protein), supporting the hypothesis that matrilin-3 targeting allows IGF-1–ctAb to remain in proximity to the cell surface for a longer period of time, extending the duration of IGF-1 receptor activation [Reference 5]. We also recently studied fusion proteins that were re-engineered from a monomeric structure into a dimeric IgG–like antibody structure. This IgG–like IGF-1 fusion protein showed an extended serum half-life, stimulated long bone growth, and did not induce hypoglycemia.
Additional Funding
- CRADA with Cavalry Biosciences
- NICHD Early Career Award
- NICHD Career Development Award
Publications
- A neomorphic variant in the transcription factor SP7 alters sequence specificity and causes a high-turnover bone disorder. Nat Commun 2022 13:700
- Variants in WASHC3, a component of the WASH complex, cause short stature, variable neurodevelopmental abnormalities, and distinctive facial dysmorphism. Genet Med Open 2024 3:101915
- Epigenetic causes of overgrowth syndromes. J Clin Endocrinol Metab 2024 109:312-332
- Loss of function variant in SPIN4 causes an X-linked overgrowth syndrome. JCI Insight 2023 8(9):e167074
- Efficacy of cartilage-targeted IGF-1 in a mouse model of growth hormone insensitivity. Front Endocrinol (Lausanne) 2025 15:1523931
Collaborators
- Lijin Dong, PhD, Genetic Engineering Core, NEI, NIH, Bethesda, MD
- Youn Hee Jee, MD, Children’s National Medical Center, Washington DC
- Ola Nilsson, MD, PhD, Karolinska Institute, Stockholm, Sweden
- Robert O'Brien, PhD, Cavalry Biosciences, Burlington, VT
Contact
For more information, email jeffrey.baron@nih.gov or visit https://www.nichd.nih.gov/research/atNICHD/Investigators/baron.