
Biophysics of Large Membrane Channels

- Sergey M. Bezrukov,
PhD, DSci, Head, Section on Molecular Transport - Tatiana K. Rostovtseva, PhD, Associate Scientist
- Megha Rajendran, PhD, Visiting Fellow
- Wendy Fitzgerald, BS, Biologist
- Antara Syam, BS, Graduate Partnerships Program Predoctoral Fellow
- Glenine Bautista, BS, Postbaccalaureate Fellow
- Nathan E. Wohlstadter, BS, Postbaccalaureate Fellow
- Mya Wolfe, BA, Postbaccalaureate Fellow
- Alexander M. Berezhkovskii, PhD, Contractor
We study the functional properties and regulation of beta-barrel channels, a subgroup of transmembrane proteins that form cylindrical pores composed of beta-strands. While they account for a small percentage of the total proteome, most integral transmembrane proteins in the outer membrane of mitochondria, Gram-negative bacteria, and chloroplasts are beta-barrel proteins. Gram-positive bacteria do not have integral beta-barrel transmembrane proteins; however, they secrete beta-barrel toxins such as anthrax toxin PA63 (Bacillus anthracis) and alpha-hemolysin (Staphylococcus aureus). Beta-barrel channels of Gram-negative bacteria transport ions (OmpF), metabolites (LamB), and proteins (BamA). Sam50, a mitochondrial ortholog of BamA, is an even-numbered beta-barrel that can be traced to a common bacterial ancestor. However, two other mitochondrial outer membrane proteins, Tom40 and VDAC, composed of 19 beta-strands, likely evolved independently in early mitochondria. While Tom40 complements Sam50 in transporting and assembling mitochondrial proteins, VDAC regulates the exchange of ions and metabolites between the cytosol and mitochondria. Given its location at the interface between the cytosol and mitochondria, VDAC serves as a cellular hub that governs calcium signaling, cell metabolism, and cell death. Predictably, VDAC has been implicated in many diseases such as cancer, diabetes, and heart and neurodegenerative diseases, making it a promising pharmacological target.
Our lab combines methods of theoretical physics with biophysical and cell-biology techniques to elucidate the physical principles of channel regulation under normal and pathological conditions. We investigate the properties of these channels by functional reconstitution of the channel-forming proteins into planar lipid membranes. This benchmark technique, i.e., single-channel electrophysiology, enables us to study the effects of lipids, proteins, and drugs under well defined conditions at single-molecule resolution. We complement this technique by other biophysical methods, such as fluorescence-correlation spectroscopy, bilayer-overtone analysis, and quartz-crystal microbalance dissipation monitoring to study the effect of proteins and drugs on membranes. We recently expanded our capabilities to include cell-biology methods such as flow cytometry, Seahorse (extracellular flux assay) real-time metabolic assays, and microscopy. Together, these methods advance our understanding of the precise molecular mechanisms of channel regulation by protein interactome and drugs.
The combination of the methods mentioned above has led to the identification and molecular characterization of VDAC complexation with cytosolic proteins such as tubulin and alpha-synuclein. We hope that the discovery of these complexations and their effects on mitochondrial function in live cells will bolster the development of pharmacological approaches to effectively correct the deviant molecular interactions associated with pathology in chronic and acute diseases and in development.
Global and local effects in lipid-mediated interactions between peripheral and integral membrane proteins
Amphitropic proteins (APs) are a subfamily of water-soluble peripherally membrane-bound proteins that interact directly with the lipid membrane rather than with intrinsic membrane proteins. They are therefore strongly influenced by the properties of the membrane. The notion that lipids are not merely passive fillers of the space between membrane proteins, but actively interact with them, controlling protein conformational transitions and impacting their function, is now widely accepted. When an AP interacts with a membrane containing an integral membrane protein, a ternary protein-lipid-protein system is created. Even in the absence of direct interactions between the amphitropic and integral proteins, the two proteins can affect each other by modifying lipid membrane properties, either at the global (i.e., whole-membrane) or local (i.e., confined to a small area around the bound or integrated protein) scale. These lipid-mediated protein-protein interactions are indirect and therefore difficult to elucidate; independent experimental data are required to report on each individual interaction in order to understand the entire system. Examples for which comprehensive data are available are remarkably rare. In a recent study, we describe how these difficulties could be surmounted by using the channel-forming integral membrane protein gramicidin A (grA) reconstituted in a planar lipid membrane and exposed to the amphitropic proteins dimeric tubulin or α-synuclein. Importantly, there are no known direct interactions between these APs and grA; therefore, the changes in grA lifetime and conductance reveal the changes in the lipid membrane properties. We leverage our detailed understanding of the tubulin-lipid interaction mechanism, as well as the self-reporting properties of the grA channel, to understand how tubulin affects the properties of the lipid membrane. In this context, grA serves a dual role: first, it reports on the global properties of the lipid membrane; grA results, combined with the well understood tubulin-lipid interaction, yield a complete picture of the mutual effect of tubulin binding on the lipid membrane. Second, the presence of the grA–conducting dimer alters the local membrane curvature and creates binding sites for tubulin in an otherwise inert membrane composition. Similarly, we also found that α-synuclein increases grA channel lifetime in a dose-dependent manner. The practical importance of our study is based on the observation that many membrane-altering small molecules, such as anesthetics, tricyclic antidepressants, and psychedelics, have membrane-modifying properties similar to APs. Thus, the recognition of lipid-mediated protein-protein interactions could be instrumental in understanding the off-target effects of some of these drugs on cell and organelle membranes, and therefore on membrane proteins residing in, or peripherally associated with, those membranes.
Figure 1. Lipid-mediated protein-protein interactions between an amphitropic protein, dimeric tubulin, and an integral membrane protein, gramicidin A (grA)
Both global and local membrane properties play a role and are reported on by the lifetime and conductance of the grA channel, respectively. Block arrows denote a direction of influence and do not indicate chemical equilibrium. Sections of Reference 1, in which the various interactions are discussed, are denoted by §. The lipid influence on the grA channel illustrated in the upper right corner of the cartoon was shown to be reciprocal. However, the oppositely directed effect occurs only at much higher gramicidin concentrations of one molar percent or higher, and is thus not relevant to our study. Created in BioRender.com.
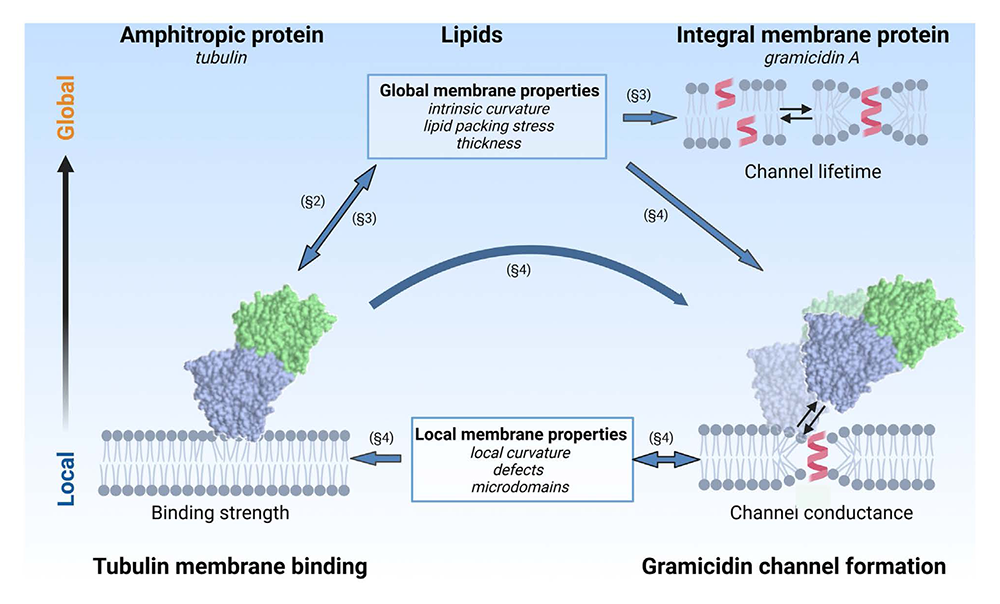
Figure 1. Lipid-mediated protein-protein interactions between an amphitropic protein, dimeric tubulin, and an integral membrane protein, gramicidin A (grA)
Both global and local membrane properties play a role and are reported on by the lifetime and conductance of the grA channel, respectively. Block arrows denote a direction of influence and do not indicate chemical equilibrium. Sections of Reference 1, in which the various interactions are discussed, are denoted by §. The lipid influence on the grA channel illustrated in the upper right corner of the cartoon was shown to be reciprocal. However, the oppositely directed effect occurs only at much higher gramicidin concentrations of one molar percent or higher, and is thus not relevant to our study. Created in BioRender.com.
Conformational plasticity of mitochondrial VDAC2 controls the kinetics of its interaction with cytosolic proteins.
The voltage-dependent anion channel (VDAC) is the most abundant integral protein of the mitochondrial outer membrane. It is the major pathway for water-soluble metabolites and small ions to cross this membrane. In mammals, there are three isoforms of the VDAC protein: VDAC1, VDAC2, and VDAC3. Despite approximately 70% sequence similarity between the isoforms and their ability to form large conductive anionic channels (about 4 nS in 1 M KCl at room temperature), which gate in response to the applied voltage when reconstituted into a planar lipid membrane, each VDAC isoform has a distinct physiological role. Among the three isoforms, VDAC2 is unique because of its embryonic lethality upon knockout. Using single-molecule electrophysiology, we investigated the biophysical properties that distinguish VDAC2 from VDAC1 and VDAC3. We found that, unlike the latter, VDAC2 exhibits dynamic switching between multiple high-conductance, anion-selective substates. Using α-synuclein (αSyn), a known VDAC1 cytosolic regulator, we established that higher-conductance substates correlate with increased on-rates of αSyn–VDAC2 interaction but shorter blockage times, thus maintaining an unchanged equilibrium constant across all substates. This suggests that αSyn detects VDAC2’s structural variations before the final step in the binding reaction. We explored the dependence of VDAC2’s unique amino-terminal extension and cysteines on the substate behavior, finding that both structural elements modulate the occurrence of substates. The discovered conformational flexibility of VDAC2 may enable its interactions with diverse binding partners, explaining its critical physiological role via dynamic adaptation to cellular needs. Specifically, we propose that the appearance of distinct substates within the same channel, along with their different kinetics of interaction with αSyn, reveals the unique structural plasticity of VDAC2. This feature suggests a key to understanding the exceptional role of this multifaceted channel in the cell. Tentatively, it could explain the physiological significance of VDAC2: its ability to adapt to cell conditions and change the rates of interaction with its many cytosolic protein partners.
Figure 2. VDAC2 dynamic substates differ from one another in their interaction with the cytosolic regulator α-synuclein (αSyn)
(A) Representative current record of a single VDAC2 WT channel in the presence of 10 nM αSyn, displaying robust substate behavior manifested as spontaneous fluctuations between the low-conductance (3 nS) long-lasting (O1) and high-conductance (3.6 nS) short-lasting (O2) substates at 10 and 30 mV. After 10 min, spontaneous transition to a higher-conductance (3.8 nS) substate (O3) is observed. Insets (I) and (II) show αSyn–induced blockage events at a finer timescale for each of the three substates at 30 mV (gray boxes). Open states (O1, O2, and O3) are indicated by dashed lines, and αSyn–blocked states (B1, B2, and B3) are indicated by dotted lines. (B) The cartoon illustrating the αSyn–VDAC interaction events is characterized by the time when the channel is open (τon) and the blockage time (τb), the duration of each blockage event. (C) Representative log-binned histograms of τon [(C), i] and τb [(C), ii] of each state at 30 mV obtained from the experiment in (A). Solid lines are fits to a single-exponential function with characteristic times <τon> equal to 19.7, 8.62, and 3.87 ms for states O1, O2, and O3, respectively [(C), i]; and τoff = <τb> equal to 5.79, 2.59, and 1.63 ms for states B1, B2, and B3, respectively [(C), ii]. (D) Comparison of kinetic parameters: kon = 1/(<τon>[C]), where [C] is the αSyn bulk concentration, τoff, and the equilibrium constant, Keq = konτoff, of αSyn–VDAC binding for each state at 30 mV. Error bars are ±SD of three fitting algorithms for the exponential fits. (E) Comparison of αSyn–blocked conductance of each conducting state. ΔG/G is the relative conductance drop, where ΔG is the difference between open-state (G) and blocked-state conductances of states O1, O2, and O3. Error bars are ±SD between measurements of ΔG/G.
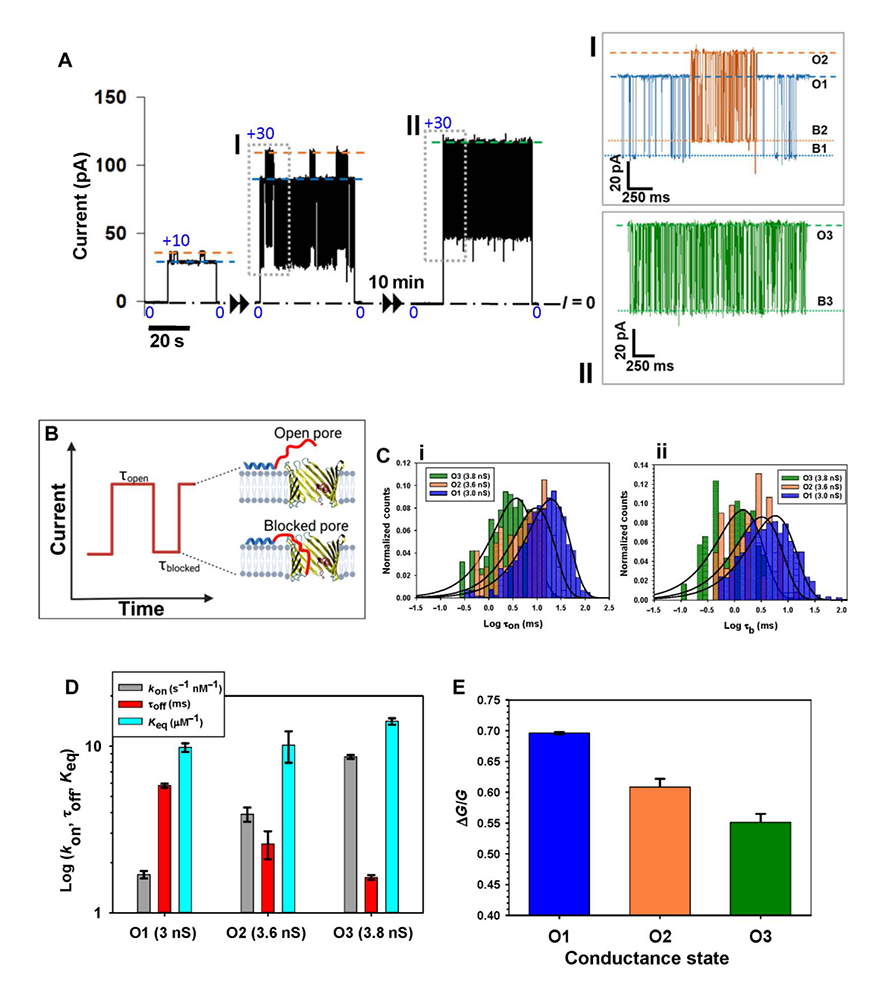
Figure 2. VDAC2 dynamic substates differ from one another in their interaction with the cytosolic regulator α-synuclein (αSyn)
(A) Representative current record of a single VDAC2 WT channel in the presence of 10 nM αSyn, displaying robust substate behavior manifested as spontaneous fluctuations between the low-conductance (3 nS) long-lasting (O1) and high-conductance (3.6 nS) short-lasting (O2) substates at 10 and 30 mV. After 10 min, spontaneous transition to a higher-conductance (3.8 nS) substate (O3) is observed. Insets (I) and (II) show αSyn–induced blockage events at a finer timescale for each of the three substates at 30 mV (gray boxes). Open states (O1, O2, and O3) are indicated by dashed lines, and αSyn–blocked states (B1, B2, and B3) are indicated by dotted lines. (B) The cartoon illustrating the αSyn–VDAC interaction events is characterized by the time when the channel is open (τon) and the blockage time (τb), the duration of each blockage event. (C) Representative log-binned histograms of τon [(C), i] and τb [(C), ii] of each state at 30 mV obtained from the experiment in (A). Solid lines are fits to a single-exponential function with characteristic times <τon> equal to 19.7, 8.62, and 3.87 ms for states O1, O2, and O3, respectively [(C), i]; and τoff = <τb> equal to 5.79, 2.59, and 1.63 ms for states B1, B2, and B3, respectively [(C), ii]. (D) Comparison of kinetic parameters: kon = 1/(<τon>[C]), where [C] is the αSyn bulk concentration, τoff, and the equilibrium constant, Keq = konτoff, of αSyn–VDAC binding for each state at 30 mV. Error bars are ±SD of three fitting algorithms for the exponential fits. (E) Comparison of αSyn–blocked conductance of each conducting state. ΔG/G is the relative conductance drop, where ΔG is the difference between open-state (G) and blocked-state conductances of states O1, O2, and O3. Error bars are ±SD between measurements of ΔG/G.
Particle dynamics in biconical cavities: first-passage, direct-transit, and looping time distributions; theoretical analysis in the framework of a continuous diffusion model
Earlier, we analyzed the effects of monotonically changing entropy potentials imposed by expanding or narrowing tubes on particle diffusion. This year, we examined particle dynamics in biconical cavities, where particle motion is influenced by either an entropy potential well, as in a cavity composed of first-expanding and then-narrowing cones, or an entropy potential barrier, as in a cavity made up of first-narrowing and then-expanding identical cones. Both types of cavities are relevant to multiple technological and biological problems, with examples of such structures found at both the micro- and nanoscales. We derive analytical expressions for the Laplace transforms of the distributions for the first-passage, direct-transit, and looping times in such structures. We found that not only the average values but also the distributions of the first-passage times in both cavities are identical. However, the direct-transit and looping time distributions are drastically different. In particular, the mean direct-transit time for the expanding-narrowing cavity (entropy potential well) approaches a constant value with the increasing depth of the entropy potential well. This corresponds to the increasing ratio of the cavity’s largest radius to the radius of its openings. In contrast, the mean direct-transit time goes to infinity in the case of the narrowing-expanding cavity with the increasing height of the entropy potential barrier. Importantly, in various single-molecule experiments, it has been demonstrated that the first passage time distributions, not just their mean values, convey crucial information about the molecular intermediates governing the dynamics of complex chemical and biological systems. In our study, we demonstrated that the distributions of looping and direct transit times can reveal the drastic differences in system geometry, even when the systems’ first passage time averages and distributions are identical.
Figure 3. Schematic representation of the types of molecule trajectories in three-dimensional biconical cavities
Cavities with the entropy potential barrier are shown in panels (a) and (b); cavities with the entropy well are shown in panels (c) and (d). Panels (a) and (c) illustrate single-loop trajectories. Panels (b) and (d) provide illustrations for the direct-transit trajectories.
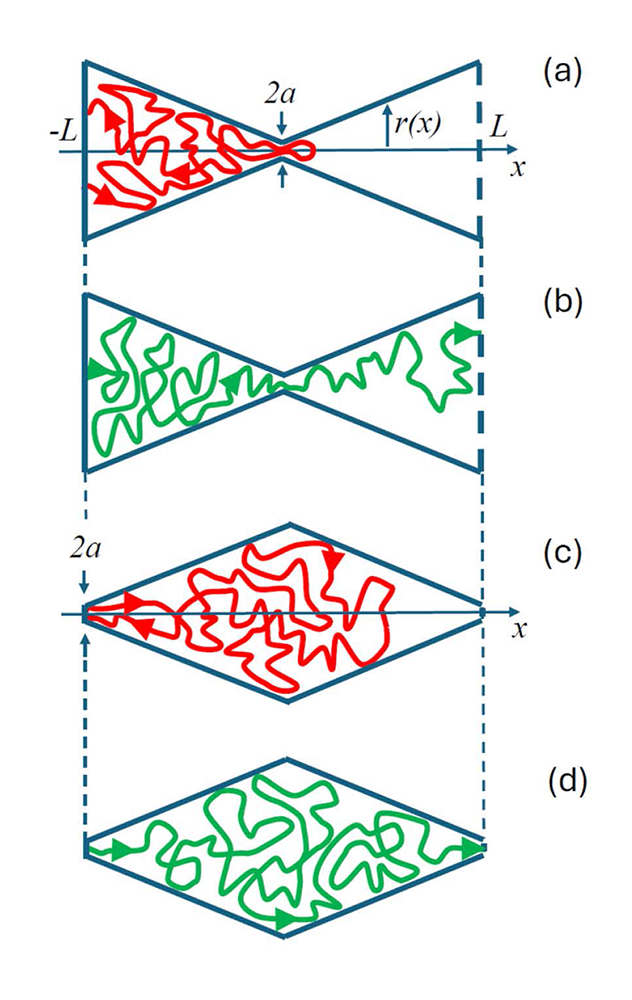
Figure 3. Schematic representation of the types of molecule trajectories in three-dimensional biconical cavities
Cavities with the entropy potential barrier are shown in panels (a) and (b); cavities with the entropy well are shown in panels (c) and (d). Panels (a) and (c) illustrate single-loop trajectories. Panels (b) and (d) provide illustrations for the direct-transit trajectories.
Additional Funding
- Graduate Partnerships Program for Antara Syam
Publications
- Global and local effects in lipid-mediated interactions between peripheral and integral membrane proteins. Front Mol Biosci 2025 12:1605772
- Conformational plasticity of mitochondrial VDAC2 controls the kinetics of its interaction with cytosolic proteins. Sci Adv 2025 11:eadv4410
- Flux through membrane channel: Linear transport vs single-molecule approaches. Phys Chem Chem Phys 2025 27:2192-2196
- Antimicrobial peptide class that forms discrete β-barrel stable pores anchored by transmembrane helices. Nat Commun 2025 16:7231
- Particle dynamics in biconical cavities: First-passage, direct-transit, and looping time distributions. J Chem Phys 2025 162:244117
Collaborators
- Vicente M. Aguilella, PhD, Universidad Jaume I, Castellón, Spain
- Leonardo Dagdug, PhD, Universidad Autónoma Metropolitana-Iztapalapa, Mexico City, Mexico
- David Hoogerheide, PhD, National Institute of Standards and Technology, Gaithersburg, MD
- Ekaterina M. Nestorovich, PhD, The Catholic University of America, Washington, DC
- William M. Rosencrans, BS, California Institute of Technology, Pasadena, CA
- Alexander Sodt, PhD, Unit on Membrane Chemical Physics, NICHD, Bethesda, MD
- Michael Weinrich, MD, Office of the Director, NICHD, Bethesda, MD
- Joshua Zimmerberg, MD, PhD, Section on Integrative Biophysics, NICHD, Bethesda, MD
Contact
For more information, email bezrukos@mail.nih.gov or visit https://www.nichd.nih.gov/research/atNICHD/Investigators/Bezrukov.