
Protein Sorting in the Endomembrane System

- Juan S. Bonifacino,
PhD, Head, Section on Intracellular Protein Trafficking - Raffaella De Pace, PhD, Staff Scientist
- Adriana Golding, PhD, Research Fellow
- William Huffman, MS, Technician
- Xiaolin Zhu, Technician
- Nireekshit Addanki Tirumala, PhD, Visiting Fellow
- Felipe Del Valle Batalla, PhD, Visiting Fellow
- Saikat Ghosh, PhD, Visiting Fellow
- Zhaoxing Ran, PhD, Visiting Fellow
- Ganesh Shelke, PhD, Visiting Fellow
- Surbhi Verma, PhD, Visiting Fellow
- Sabina Yeasmin, PhD, Visiting Fellow
- Jennifer M. Kunselman, PhD, Intramural Research Training Award Fellow
- Rafael Mattera, PhD, Special Volunteer
Our laboratory studies the molecular mechanisms underlying the sorting of transmembrane proteins (known as cargo) to various compartments within the endomembrane system of eukaryotic cells. In polarized cells such as epithelial cells and neurons, the system comprises an array of membrane-enclosed organelles, including the endoplasmic reticulum (ER), the Golgi apparatus, the trans-Golgi network (TGN), endosomes, lysosomes, lysosome-related organelles (LROs, e.g., melanosomes, cytotoxic granules), and various domains of the plasma membrane. The transport of cargo between these compartments is mediated by vesicular or tubular carriers that bud from a donor compartment, translocate through the cytoplasm, and fuse with an acceptor compartment. We study the molecular machineries that mediate these processes in the context of different intracellular transport pathways. We also investigate the mechanisms involved in organelle transport within non-polarized cells and neurons. Our fundamental research serves as a basis for explaining the pathogenetic mechanisms of protein- and organelle-transport disorders, including the pigmentation and bleeding disorder Hermansky-Pudlak syndrome (HPS), hereditary spastic paraplegias (HSPs), and other neurodevelopmental disorders.
EIPR1 variants cause a novel neurodevelopmental disorder with endolysosomal and dense-core vesicle defects.
EIPR1 (EARP–interacting protein 1, formerly TSSC1) is a WD40–domain protein (WD40 domains act as protein-interaction scaffolds) involved in delivering endosome-derived transmembrane cargos to the plasma membrane and the trans-Golgi network (TGN) and in the biogenesis of dense core vesicles. While in cultured cells and model organisms its role was known, its significance in humans was unclear. We identified five homozygous EIPR1 missense variants in eight individuals with a neurological disorder featuring neurodevelopmental delay, microcephaly, ataxia, and spasticity. These variants reduce EIPR1 protein levels and its interaction with EARP and GARP complexes (Golgi-assisted retrograde protein complexes), impairing endosomal recycling and dense-core vesicle biogenesis. Patient fibroblasts exhibit phenotypes consistent with EARP and GARP deficiencies. Knockout of eipr1 in zebrafish causes defects that are rescued by wild-type human EIPR1 mRNA but not by variant mRNAs, confirming impaired activity. These findings link EIPR1 to a neurodevelopmental disorder and highlight its essential role in endosomal recycling and vesicle biogenesis.
The hereditary spastic paraplegia type 21 (SPG21) protein is a RAB7A effector that promotes noncanonical mTORC1 signaling.
Hereditary spastic paraplegia type 21 (SPG21) is an inherited neurological disorder caused by biallelic mutations in the SPG21 gene, which encodes the protein maspardin. We found that the SPG21 protein localizes to endolysosomes via its interaction with the GTP–bound form of RAB7A (protein that regulates endolysosomal trafficking). Disease-associated SPG21 variants reduce SPG21 expression and disrupt its endolysosomal localization in both non-neuronal cells and neurons. Functional analysis connects SPG21 to endolysosomal and mTORC1 (a protein kinase complex that functions as a nutrient/energy/redox sensor and controls protein synthesis) signaling pathways. Biochemical studies show that SPG21 depletion does not affect the phosphorylation of canonical mTORC1 substrates such as ULK1, S6K1, and 4E-BP1, but reduces the phosphorylation of the non-canonical mTORC1 substrate TFEB, a central regulator of the autophagy/lysosomal-to-nucleus signaling pathway. This reduction enhances TFEB's nuclear localization and the expression of specific TFEB–target genes. Therefore, SPG21 acts as a RAB7A effector that promotes non-canonical mTORC1–mediated phosphorylation of TFEB, suppressing its nuclear localization and transcriptional activity. These findings link SPG21 dysfunction to altered endolysosomal signaling, providing new insights into SPG21 pathogenesis.
Biallelic BORCS5 and BLOC1S1 variants cause an infantile-onset neurodegenerative disorder with altered lysosome dynamics.
The BLOC-one–related complex (BORC) is a multiprotein complex involved in positioning of the lysosome within the cytoplasm, which is composed of eight subunits, named BORCS1 (also known as BLOC1S1) through BORCS8. BORC associates with the cytosolic surface of lysosomes, where it facilitates the recruitment of the small GTPase ARL8 and kinesin-1 and kinesin-3 microtubule motors. Such recruitment promotes the anterograde transport of lysosomes toward the peripheral cytoplasm in non-neuronal cells and toward distal axons in neurons. In previous work, we demonstrated that biallelic variants in BORCS8 cause an early-infantile neurodegenerative disorder. Over the past year, through two independent collaborations, one with Niccolò Mencacci and colleagues and another with Adeline Vanderver and colleagues, we identified additional patients with biallelic variants in two other BORC subunits: BORCS5 and BLOC1S1. These patients, all children, presented with a spectrum of neurodevelopmental abnormalities, including global developmental delay, severe to profound intellectual disability, hypotonia, limb spasticity, muscle wasting, dysmorphic facial features, optic atrophy, leuko-axonopathy with hypomyelination, and other neurodegenerative features predominantly affecting supratentorial brain regions. Cellular studies revealed that the pathogenic variants in BORCS5 and BLOC1S1 result in reduced protein expression, impaired assembly with other BORC subunits, and/or compromised ability to mediate lysosome translocation to the cell periphery. Together, these findings establish BORCS5 and BLOC1S1 as novel genetic loci for early-infantile neurodegenerative disorders and underscore the essential role of BORC and lysosomal dynamics in the development and maintenance of the central nervous system.
Figure 1. Schematic representation of the function of the BORC complex in the movement of lysosomes and related organelles
Abbreviations: BORC: BLOC-1–related complex, a lysosome motility and fusion regulator; ARL8: ARF-like 8 small GTPase; PLEKHM2: Pleckstrin homology and RUN domain containing M2 protein, a lysosome-kinesin adaptor; KIF1/UNC-104: kinesin-3 motor; SVP: synaptic vesicle precursor; DCV: dense-core vesicle; RUFY3/4: ARL8–effector proteins that regulate coupling of lysosomes to dynein-dynactin; HOPS: homotypic fusion and vacuole-protein sorting complex; RAB7/2: small GTPases that regulate endolysosomal trafficking; WT: wild-type; KO: knockout
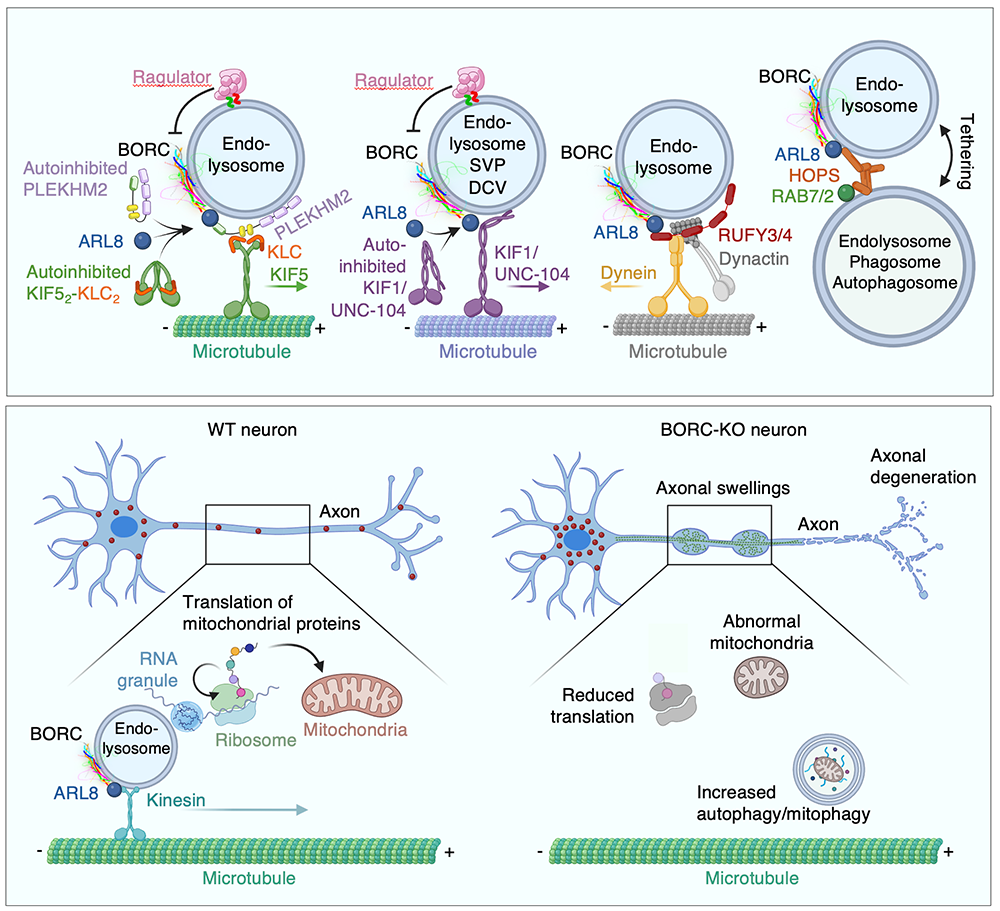
Figure 1. Schematic representation of the function of the BORC complex in the movement of lysosomes and related organelles
Abbreviations: BORC: BLOC-1–related complex, a lysosome motility and fusion regulator; ARL8: ARF-like 8 small GTPase; PLEKHM2: Pleckstrin homology and RUN domain containing M2 protein, a lysosome-kinesin adaptor; KIF1/UNC-104: kinesin-3 motor; SVP: synaptic vesicle precursor; DCV: dense-core vesicle; RUFY3/4: ARL8–effector proteins that regulate coupling of lysosomes to dynein-dynactin; HOPS: homotypic fusion and vacuole-protein sorting complex; RAB7/2: small GTPases that regulate endolysosomal trafficking; WT: wild-type; KO: knockout
Programmed ribosomal frameshifting during mRNA decoding generates a constitutively active PLEKHM2 proteoform.
In collaboration with Yousuf Khan and other colleagues, we discovered a novel mechanism by which cells generate a proteoform of the lysosome-kinesin adaptor PLEKHM2 (also known as SKIP) through programmed ribosomal frameshifting. The process involves a subset of ribosomes that alter their reading frame on an mRNA. Although frameshifting is commonly utilized by viruses, there are very few known phylogenetically conserved examples in nuclear-encoded genes. We identified a +1 frameshifting event during the decoding of the human gene PLEKHM2, which encodes an essential adaptor for lysosomal trafficking, an event that grants access to a second, internally overlapping open reading frame. The resulting carboxyl-terminal domain of this frameshift protein forms an α-helix, releasing PLEKHM2 from autoinhibition and enabling its movement to the tips of cells independently of activation by the GTPase ARL8
A SYS1–ARFRP1–ARL5–ARMH3–PI4KB axis regulates PI4P synthesis at the TGN.
ARL5 is a small GTPase of the ARF family that is recruited to the TGN by the ARF–family GTPase ARFRP1, in complex with the transmembrane protein SYS1. At the TGN, ARL5 mediates the recruitment of its known effector, the GARP–tethering complex, promoting SNARE–dependent fusion of endosome-derived retrograde transport carriers with the TGN. To explore additional functions of ARL5, we performed proximity biotinylation and protein-interaction assays to identify novel effectors. These analyses revealed that the armadillo-repeat protein (a family of proteins that contain several tandemly repeated copies composed of a pair of alpha helices that form a hairpin structure) ARMH3 (C10orf76) specifically binds to the active, GTP–bound form of ARL5 and is recruited to the TGN in a manner dependent on SYS1, ARFRP1, and ARL5. Unlike GARP, ARMH3 is not required for retrograde cargo transport. Instead, ARMH3 plays a distinct role by activating phosphatidylinositol 4-kinase III beta (PI4KB), which generates the major pool of phosphatidylinositol 4-phosphate (PI4P) at the TGN. This PI4P pool supports the recruitment of the oncoprotein GOLPH3 and facilitates glycan processing at the TGN. Collectively, these findings identify a novel SYS1–ARFRP1–ARL5–ARMH3 axis that regulates PI4KB–mediated PI4P production at the TGN, expanding our understanding of lipid signaling and membrane trafficking at this organelle.
Messenger RNA transport on lysosomal vesicles maintains axonal mitochondrial homeostasis and prevents axonal degeneration.
In neurons, RNA granules are transported along the axon for local translation away from the soma. Recent studies indicate that some of this transport involves the hitchhiking of RNA granules on lysosome-related vesicles. To identify a subset of axonal mRNAs that depend on lysosome-related vesicles for transport, this past year we leveraged the ability to prevent transport of these vesicles into the axon by knocking out the lysosome-kinesin adaptor BLOC-one–related complex (BORC), which we had discovered in previous research. We found that BORC knockout causes depletion of a large group of axonal mRNAs that mainly encode ribosomal and mitochondrial/oxidative phosphorylation proteins. The depletion results in mitochondrial defects and eventually leads to axonal degeneration in human induced pluripotent stem cell (iPSC)–derived and mouse neurons. Pathway analyses of the depleted mRNAs revealed a mechanistic connection between BORC deficiency and common neurodegenerative disorders. The findings demonstrate that mRNA transport on lysosome-related vesicles is critical for the maintenance of axonal homeostasis and that its failure causes axonal degeneration.
Publications
- EIPR1 variants cause a neurodevelopmental disorder with endolysosomal and dense core vesicle defects. Brain 2025 doi:10.1093/brain/awaf371
- Programmed ribosomal frameshifting during PLEKHM2 mRNA decoding generates a constitutively active proteoform that supports myocardial function. Sci Adv 2025 11:eady1742
- Messenger RNA transport on lysosomal vesicles maintains axonal mitochondrial homeostasis and prevents axonal degeneration. Nat Neurosci 2024 27:1087-1102
- ARMH3 is an ARL5 effector that promotes PI4KB-catalyzed PI4P synthesis at the trans-Golgi network. Nat Commun 2024 15:10168
- Biallelic BORCS8 variants cause an infantile-onset neurodegenerative disorder with altered lysosome dynamics. Brain 2024 147:1751-1767
Collaborators
- Tamas Balla, MD, PhD, Section on Molecular Signal Transduction, NICHD, Bethesda, MD
- Ryan K. Dale, MS, PhD, Bioinformatics and Scientific Programming Core, NICHD, Bethesda, MD
- Yousuf Khan, PhD, Stanford University, Stanford, CA
- Reza Maroofian, PhD, University College London, London, United Kingdom
- Niccolo Mencacci, MD, PhD, Northwestern University, Chicago, IL
- Nicole Y. Morgan, PhD, Center for Biomedical Engineering Technology Acceleration (BETA), NIBIB, Bethesda, MD
- Shunmoogum A. Patten, PhD, INRS-Institut Armand Frappier, Laval, Canada
- Adeline Vanderver, MD, Children's Hospital of Philadelphia Research Institute, Philadelphia, PA
- Michael E. Ward, MD, PhD, Inherited Neurodegenerative Diseases Unit, NINDS, Bethesda, MD
Contact
For more information, email juan.bonifacino@nih.gov or visit https://www.nichd.nih.gov/research/atNICHD/Investigators/bonifacino.