
Mechanisms of Membrane Fusion in the Formation of Multinucleated Osteoclasts and Myotubes

- Leonid V. Chernomordik,
PhD, Head, Section on Membrane Biology - Eugenia Leikina, DVM, Senior Research Assistant
- Kamram Melikov, PhD, Staff Scientist
- Elena Zaitseva, PhD, Staff Scientist
- Griffin Katz, BS, Postbaccalaureate Fellow
- Katherine Kennedy, BS, Postbaccalaureate Fellow
- Rachel McCarley, BS, Postbaccalaureate Fellow
- Alexandra Swisher, BS, Postbaccalaureate
Diverse biological processes, in which enveloped viruses infect cells, and cells from all kingdoms of life secrete, internalize, traffic and sort integral proteins, sculpt their membranes, and bring together parent genomes in sexual reproduction, share a common stage: fusion of two membranes into one. Biological membrane remodeling is tightly controlled by protein machinery, but is also dependent on the lipid composition of the membranes. Whereas each kind of protein has its own individual personality, membrane lipid bilayers have rather general properties, manifested by their resistance to disruption and bending, and by their charge. Our long-term goal is to understand how proteins fuse membrane lipid bilayers. We expect that better understanding of important fusion reactions will bring about new ways of controlling them and lead to new strategies for quelling diseases involving cell invasion by enveloped viruses and defects in intracellular trafficking or intercellular fusion. Our general strategy is to combine in-depth analysis of the best characterized fusion reactions with comparative analysis of diverse, less explored fusion reactions, which can reveal new kinds of fusion proteins and clarify the generality of emerging mechanistic insights. In our recent studies, we explored the role of calcium and lipid signaling in the formation of multinucleated osteoclasts and the role of ether lipids in myoblast fusion.
Phosphatidylserine exposure and annexin A5 weaken the actin cortex in osteoclast fusion.
Cell-cell fusion plays a critical role in fertilization, the lifelong remodeling of bones and skeletal muscles, the formation of the placental syncytiotrophoblast, and other physiological processes. Some mechanistic motifs appear to be strikingly conserved across these diverse cell-cell fusion processes. All listed cell-cell fusions depend on lipid scramblases, proteins that mediate the nonselective, bidirectional movement of lipids between the inner (cytofacial) and outer leaflets of the plasma membrane (PM). All fusions depend on scramblase-mediated non-apoptotic cell-surface exposure of an anionic lipid phosphatidylserine (PS), normally almost exclusively retained in the inner leaflet of the PM. These fusions also depend on Annexin A5 (Anx A5).
Membrane fusion depends on membrane deformations, and initiation of PM deformations depends on the local and transient detachment of a patch of membrane from the actin cortex (AC), the membrane-linked actin-rich protein network. The AC is attached to the inner leaflet of the PM via membrane-anchoring proteins of the ezrin/radixin/moesin (ERM) family. ERM binding to the PM and organization of the actomyosin network depend on the membrane lipid composition and are promoted by PS and another anionic lipid, phosphatidylinositol 4,5-bisphosphate [PI(4,5)P2]. Like PS, PI(4,5)P2 is highly enriched in the inner leaflet of PM, but in some physiological processes it is also found at the cell surface. Local weakening of the AC–PM attachment, via mechanisms such as reduction of the negative surface charge of the inner leaflet, allows intracellular pressure to locally bulge the PM.
Why do diverse cell fusion processes depend on lipid scramblase activity and on the appearance of PS and Anx A5 at the cell surface? In our most recent study [Reference 2], we address this question for osteoclast fusion, a critical stage in the formation of bone-resorbing, multinucleated osteoclasts. Each fusion event increases the bone-resorbing activity of osteoclasts, which balances the bone-forming activity of osteoblasts in the tightly regulated, lifelong remodeling of the skeleton. We found that the cell-cell fusion stage of osteoclastogenesis depends on a rise in intracellular calcium, and that the amounts of Anx A5 at the surface of fusion-committed osteoclasts limit the efficiency of their fusion. Calcium signaling promotes lipid scrambling detected as PS exposure. Activation of lipid scrambling reduces PS and PI(4,5)P₂ levels in the inner leaflet of the PM, leading to local AC–PM detachment. Binding of extracellular Anx A5 to cell-surface PS enhances PS depletion from the inner leaflet and thus further weakens the AC–PM connection. Based on these findings and theoretical analysis, we propose that the PS exposure–Anx A5–binding–AC detachment pathway facilitates osteoclast fusion and other cell-cell fusions by promoting membrane deformations required for formation of prefusion membrane contacts. Additionally, our model suggests that a local increase in tension in the AC–detachment region of the membrane promotes fusion pore expansion. Indeed, we found that lowering PM tension by placing the cells into a hypertonic medium inhibits osteoclast fusion. Based on these findings and theoretical analysis, we propose that the Ca2+ signaling—PS exposure—Anx A5–binding—AC detachment pathway (Figure 1) facilitates osteoclast fusion and other cell-cell fusions by promoting membrane deformations required for formation of prefusion membrane contacts. Additionally, our model suggests that a local increase in tension in the AC detachment region of the membrane promotes fusion pore expansion.
Figure 1. Hypothetical pathway from intracellular Ca2+ rise to PS–exposure to fusion-promoting deformation of plasma membrane
In the initial state, the proteins of the ezrin/radixin/moesin (ERM) family (brown squares)–mediated connection between phosphatidylserine (PS) (yellow-headed lipid)–enriched inner leaflet of the membrane and underlying actin cortex AC (green lines) restricts membrane deformations. Transition1: Lipid scramblase activated by intracellular Ca2+ rise releases anionic lipids, including PS, from the inner to outer leaflet of the membrane, which results in a local detachment of a fraction of ERM and actin cortex from the plasma membrane (PM). Transition 2: Extracellular annexin A5 (Anx A5) binds to cell-surface PS and further reduces PS content in the inner membrane leaflet and ERM and cortex attachment. Transition 3: Bulging of the membrane region disconnected from actin cortex and local increase in tension in this membrane region facilitate closer approach between fusing membranes and drive fusion-pore formation [Golani G, Leikina E et al. Nat Commun 2021;12:495].
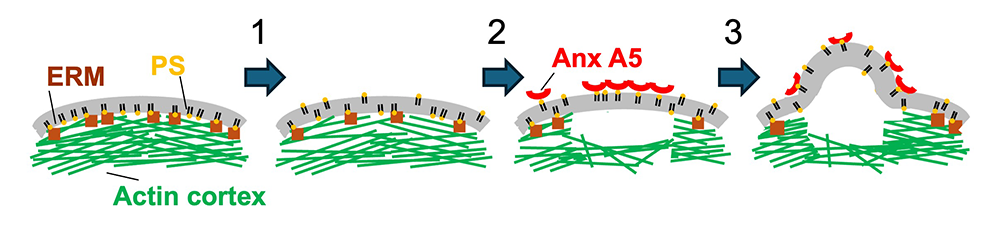
Figure 1. Hypothetical pathway from intracellular Ca2+ rise to PS–exposure to fusion-promoting deformation of plasma membrane
In the initial state, the proteins of the ezrin/radixin/moesin (ERM) family (brown squares)–mediated connection between phosphatidylserine (PS) (yellow-headed lipid)–enriched inner leaflet of the membrane and underlying actin cortex AC (green lines) restricts membrane deformations. Transition1: Lipid scramblase activated by intracellular Ca2+ rise releases anionic lipids, including PS, from the inner to outer leaflet of the membrane, which results in a local detachment of a fraction of ERM and actin cortex from the plasma membrane (PM). Transition 2: Extracellular annexin A5 (Anx A5) binds to cell-surface PS and further reduces PS content in the inner membrane leaflet and ERM and cortex attachment. Transition 3: Bulging of the membrane region disconnected from actin cortex and local increase in tension in this membrane region facilitate closer approach between fusing membranes and drive fusion-pore formation [Golani G, Leikina E et al. Nat Commun 2021;12:495].
Dissection of the signaling pathways that control fusion and bone-resorbing activity can help in identifying potential therapeutic targets for bone diseases. Indeed, some of the proteins involved in the early stages of osteoclastogenic differentiation have already been tested in animal and clinical studies as potential therapeutic targets. Moreover, we suggest that weakening the AC–PM attachment via lipid scrambling and Anx A5–PS interactions at the surface of the cells may, in addition to fusion, play an important role in other physiological contexts, including the immune response, neuronal regeneration and degeneration, and in cancer cells in metastases. A better understanding of the cascade of events within this emerging inside-out/outside-in signaling pathway will likely help uncover novel mechanisms that coordinate diverse aspects of cellular responses in various biological contexts.
Myomaker and ether lipids cooperate to promote fusion-competent membrane states.
Fusion of myoblasts, a key stage in the formation of multinucleated myofibers, and a fundamental process critical for skeletal muscle development and regeneration, depends on two muscle-specific proteins: Myomaker and Myomerger, which are essential for this fusion process. In our earlier work [Golani G, Leikina E, et al. Nat Commun 2021;12(1):e2202490119; Leikina E, Gamage DG, et al. Dev Cell 2018;46(6):767-780], in collaboration with Douglas Millay, the discoverer of these proteins, we found that these proteins independently mediate distinct steps in the fusion pathway, where Myomaker is involved in membrane hemifusion and Myomerger is necessary for fusion pore formation. In a recent study [Reference 5], also collaboration with Douglas Millay, we identified ether-linked phospholipids as modulators of Myomaker activity. These lipids are enriched in Myomaker-containing lentiviral particles, and an increase in their concentrations by modifications of lipid biosynthesis increases fusion of cells expressing muscle fusogens. To explore the mechanisms of the induction of fusion with ether lipids and Myomaker, we tested whether plasmalogens (glycerophospholipids with a vinyl-ether bond that play a role in cell membrane stability and cholesterol synthesis) can regulate hemifusion in myoblasts by adding exogenous plasmalogens, namely a synthetic lipid C18(Plasm)-22:6 PE 1-(1Z-octadecenyl)-2-docosahexaenoyl-sn-glycero-3-phosphoethanolamine, to either Myomaker-deficient or Myomerger-deficient myoblasts. Using a lipid mixing assay as a readout for hemifusion, we previously showed that Myomerger-deficient myoblasts can undergo hemifusion due to the presence of Myomaker [Leikina E, Gamage DG, et al. Dev Cell 2018;46(6):767-780]. Addition of plasmalogens to Myomerger-deficient myoblasts increased hemifusion but had no effect on complete fusion. The finding that adding a synthetic plasmalogen to the plasma membrane directly influences myoblast fusion supports the results of our experiments in which we modified concentrations of ether lipids by targeting their biosynthesis. Since the relative efficiencies of hemifusion and fusion-pore formation depend on compositions of the membranes, it is noteworthy that, in the context of myoblasts, plasmalogen concentrations sufficient to promote hemifusion in the presence of Myomaker did not promote hemifusion in the absence of this protein, and did not promote complete fusion in the absence of Myomerger. These data argue against the hypothesis that elevated ether lipids promote hemifusion-to-fusion transition by compensating for Myomerger’s activity, and suggest that ether lipids enhance the function of Myomaker to drive hemifusion. While in the case of Myomaker-expressing BHK cells, some hemifusion intermediates can spontaneously advance to complete fusion, in myoblasts hemifusion-to-fusion transition is mainly controlled by Myomerger. Based on this consideration, we propose that ether lipids cooperate with Myomaker rather than act on their own. Taken together with our earlier studies [Golani G, Leikina E, et al. Nat Commun 2021;12:e2202490119; Leikina E, Gamage DG, et al. Dev Cell 2018;46:767-780], our results suggest that ether lipids enhance the ability of Myomaker to form hemifusion intermediates. These hemifusions are then converted into fusion pores by Myomerger shifting the spontaneous curvature of the outer membrane leaflets to more positive values (Figure 2). Our findings establish a new link between lipid composition of plasma membranes and the molecular machinery responsible for muscle cell fusion.
Figure 2. Hypothetical pathway of Myomaker- and Myomerger-driven myoblast fusion
In emerging pathway, ether lipid–activated [Reference 5] Myomaker promotes hemifusion of plasma membranes of the myoblasts [Golani G, Leikina E et al. Nat Commun 2021;12:495; Leikina E, Gamage DG, et al. Dev Cell 2018;46:767]. Myomerger promotes fusion-pore opening by generating elastic stresses in the hemifusion diaphragm [Golani G, Leikina E et al. Nat Commun 2021;12:495].
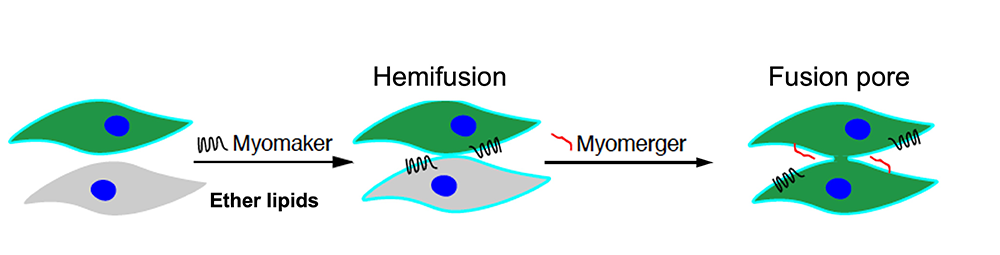
Figure 2. Hypothetical pathway of Myomaker- and Myomerger-driven myoblast fusion
In emerging pathway, ether lipid–activated [Reference 5] Myomaker promotes hemifusion of plasma membranes of the myoblasts [Golani G, Leikina E et al. Nat Commun 2021;12:495; Leikina E, Gamage DG, et al. Dev Cell 2018;46:767]. Myomerger promotes fusion-pore opening by generating elastic stresses in the hemifusion diaphragm [Golani G, Leikina E et al. Nat Commun 2021;12:495].
Additional Funding
- Binational Science Foundation (BSF) Award 2021168 (2023–2026)
- CRADA C-023-2024 with Alexion Pharmaceuticals, Inc
Publications
- Cell fusion in osteoclastogenesis. Biochem Soc Trans 2025 53:1493-1505
- Phosphatidylserine exposure and annexin A5 weaken the actin cortex in osteoclast fusion. J Cell Biol 2026 225(2):e202508055
- Cell-cell fusion: To lose one life and begin another. Bioessays 2025 47:10.1002/bies.202400206
- Elevated surface La promotes hyperfusion and contributes to impaired resorption in osteopetrosis. bioRxiv 2025 10.1101/2025.09.07.674639;preprint
- Myomaker and ether lipids cooperate to promote fusion-competent membrane states. Cell Rep 2026 45(2):116900
Collaborators
- Alison Boyce, MD, Metabolic Bone Disorders Unit, NIDCR, Bethesda, MD
- Michael Collins, MD, Skeletal Disorders & Mineral Homeostasis Section, NIDCR, Bethesda, MD
- Ari Elson, PhD, Department of Molecular Genetics, The Weizmann Institute of Science, Rehovot, Israel
- Benjamin Geiger, PhD, Department of Immunology and Regenerative Biology, The Weizmann Institute of Science, Rehovot, Israel
- Michael M. Kozlov, PhD, DHabil, Gray Faculty of Medical and Health Sciences, Tel Aviv University, Tel Aviv, Israel
- Leonid B. Margolis, PhD, Ilia University, Tiflis, Republic of Georgia
- Douglas P. Millay, PhD, Division of Molecular Cardiovascular Biology, Cincinnati Children’s Hospital Medical Center, Cincinnati, OH
- Jarred M. Whitlock, PhD, Department of Molecular Physiology and Biological Physics, University of Virginia School of Medicine, Charlottesville, VA
Contact
For more information, email chernoml@mail.nih.gov or visit https://www.nichd.nih.gov/research/atNICHD/Investigators/chernomordik.