
Molecular Genetics of Heritable Human Disorders
- Janice Y. Chou,
PhD, Head, Section on Cellular Differentiation - Irina Arnaoutova, PhD, Staff Scientist
- Lisa Zhang, PhD, Staff Scientist
- Hung-Dar Chen, PhD, Contract Technician
- Cheol Lee, PhD, Contract Technician
- Kunal Pratap, PhD, Visiting Fellow
- Ananya Samanta, PhD, Visiting Fellow
- Brian C. Mansfield, PhD, Special Volunteer

We conduct research to delineate the pathophysiology of and develop novel therapies for type I glycogen storage disease (GSD-I) subtypes GSD-Ia and GSD-Ib. GSD-Ia is caused by a deficiency in the liver/kidney/intestine-–restricted glucose-6-phosphatase-α (G6Pase-α or G6PC1) and GSD-Ib is caused by a deficiency in the ubiquitously expressed glucose-6-phosphate transporter (G6PT or SLC37A4). G6Pase-α is an endoplasmic-reticulum (ER) transmembrane protein that regulates intracellular glucose production by catalyzing the hydrolysis of G6P to glucose and phosphate. The active site of G6Pase-α faces into the ER lumen and depends on G6PT, another ER transmembrane protein, to translocate G6P from the cytoplasm into the ER lumen. To function, G6Pase-α couples with G6PT to form a G6Pase-α/G6PT complex, which maintains interprandial glucose homeostasis. GSD-Ia and GSD-Ib patients manifest a common metabolic phenotype of impaired glucose homeostasis and long-term complications of hepatocellular adenoma/carcinoma (HCA/HCC) and renal disease. There is no cure for GSD-Ia and GSD-Ib. If strictly adhered to, the current dietary therapies enable GSD-I patients to maintain a normalized metabolic phenotype. However, the underlying pathological processes remain uncorrected, and HCA/HCC and renal disease still occur in metabolically compensated GSD-I patients. We generated animal models of GSD-Ia and GSD-Ib, which are being exploited to delineate the disease more precisely and develop new treatment approaches, including gene therapy. We also generated G6PC1– and G6PT–expressing recombinant adeno-associated virus (rAAV) vectors. We showed that rAAV vector–mediated gene argumentation therapy for GSD-Ia and GSD-Ib is safe and efficacious. Our rAAV-G6PC1 vector (US patent #9,644,216) technology was licensed to Ultragenyx Pharmaceutical Inc (Novato, CA), which launched a clinical trial for GSD-Ia now entering phase III (NCT05139316). To explore alternative genetic technologies for GSD-I therapies, we have established formal collaborations under the CRADA with CRISPR Therapeutics (Cambridge, MA) and Beam Therapeutics (Cambridge, MA) to evaluate the efficacy of CRISPR/Cas9–based and adenine base editor (ABE)–based gene editing systems, respectively, to correct gene-specific G6PC1 mutations in animal models of GSD-Ia.
Figure 1. Base-editing corrects metabolic abnormalities in a humanized mouse model for glycogen storage disease type-Ia.
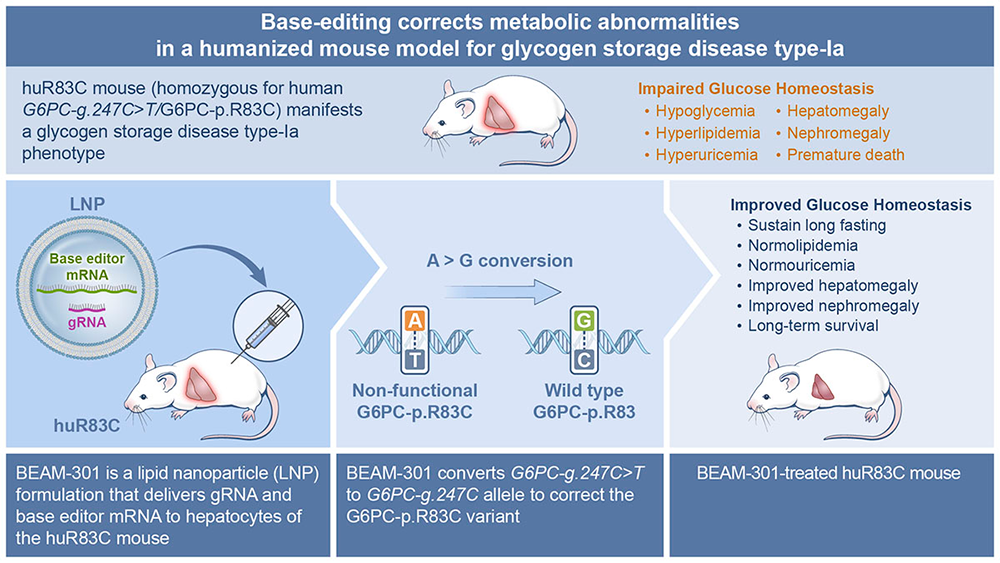
Figure 1. Base-editing corrects metabolic abnormalities in a humanized mouse model for glycogen storage disease type-Ia.
Molecular mechanism underlying hepatic autophagy impairment in GSD-Ib
The etiology of HCA/HCC in GSD-Ib is unknown. Studies have shown that deficiency in autophagy, an evolutionarily conserved, degradative process that produces energy and building blocks through lysosomal degradation of intracellular proteins and organelles in times of nutrient deprivation and environmental stress contributes to hepatocarcinogenesis. Autophagy can be regulated positively by sirtuin 1 (SIRT1), AMP–activated protein kinase (AMPK), and forkhead box O (FoxO) transcription-factor family members. In the liver, AMPK is activated via phosphorylation of the AMPK α-subunit at residue T172 by the liver kinase B-1 (LKB1), a serine/threonine kinase.
To understand the pathways contributing to hepatocarcinogenesis in GSD-Ib, we hypothesized that impaired hepatic autophagy is a significant contributor. In this study, we showed that G6PT deficiency leads to impaired hepatic autophagy, evident from attenuated expression of many components of the autophagy network, decreased autophagosome formation, and reduced autophagy flux. The G6PT–deficient liver displays impaired SIRT1 and AMPK signaling, along with reduced expression of SIRT1, FoxO3a, LKB1, and the active p-AMPK. Importantly, we showed that overexpression of either SIRT1 or LKB1 in the G6PT–deficient liver restores autophagy and SIRT1/FoxO3a and LKB1/AMPK signaling. The hepatosteatosis in the G6PT–deficient liver lowers SIRT1 expression. LKB1 overexpression reduces hepatic triglyceride levels, providing a potential link between LKB1/AMPK signaling upregulation and the increase in SIRT1 expression. In conclusion, downregulation of SIRT1/FoxO3a and LKB1/AMPK signaling underlies impaired hepatic autophagy, which may contribute to HCA/HCC development in GSD-Ib. Understanding this mechanism may guide future therapies [Reference 1].
CRISPR/Cas9–based double strand oligonucleotide insertion strategy corrects metabolic abnormalities in murine glycogen storage disease type Ia.
GSD-Ia is a pediatric genetic disorder. The rAAV-G6PC1 vector used in the Phase III clinical trial for GSD-Ia (NCT05139316) is episomally expressed. Currently, there are insufficient clinical data to understand whether multi-decade episomal transgene expression can be maintained in the human liver at a therapeutic level. We therefore explored alternative genetic technologies for GSD-Ia therapy, such as CRISPR/Cas9–based gene editing. We previously generated a G6pc-R83C mouse strain carrying the prevalent pathogenic G6PC1-p.R83C variant and showed that they exhibit the pathophysiology of impaired glucose homeostasis mimicking human GSD-Ia. In an initial exploration of CRISP/Cas-9–based editing using AAV to deliver the CRISPR reagents, we showed that a homology-directed repair strategy could correct the abnormal metabolic phenotype of neonatal G6pc-R83C mice.
Using the G6pc-R83C mice, we then explored a CRISPR/Cas9–based double-strand DNA oligonucleotide (dsODN) insertional strategy that uses the non-homologous end-joining repair mechanism to correct the pathogenic p.R83C variant in G6pc exon-2. The strategy is based on the insertion of a short dsODN into G6pc exon-2 to disrupt the native exon and to introduce an additional splice-acceptor site and the correcting sequence. When transcribed and spliced, the edited gene would generate a wild-type mRNA encoding the native G6Pase-α protein. The editing reagents formulated in lipid nanoparticles (LNP) were delivered to the liver. Mice were treated either with one dose of LNP-dsODN at age four weeks or with two doses of LNP-dsODN at age two and four weeks. The G6pc-R83C mice receiving successful editing express approximately 4% of normal hepatic G6Pase-α activity, maintain glucose homeostasis, lack hypoglycemic seizures, and display a normalized blood metabolite profile. The outcomes are consistent with preclinical studies supporting previous gene augmentation therapy, which is currently in clinical trials. Such an editing strategy may offer the basis for a therapeutic approach with an earlier clinical intervention than gene augmentation, with the additional benefit of a potentially permanent correction of the GSD-Ia phenotype [Reference 3].
Base editing corrects metabolic abnormalities and prevents hepatocarcinogenesis in murine glycogen storage disease type-Ia.
While we showed that CRISPR/Cas9–based homology-directed repair (HDR) and non-homologous end joining (NHEJ) gene-editing strategies can correct the GSD-Ia phenotype in G6pc-R83C mice, the efficacy of the HDR–mediated editing in non-dividing cells was low and the success of the NHEJ–mediated editing was dependent on the efficiency of inserting the editing DNA oligonucleotide in the correct orientation. We are therefore exploring the adenine base editor (ABE)–based technologies that enable a programmable conversion of A•T to G•C in genomic DNA for GSD-Ia therapy. The ABE system works in both dividing and non-dividing cells, is reported to produce virtually no indels or off-target editing in the genome, and can correct a pathogenic variant in its native genetic locus, leading to permanent, therapeutically effective long-term expression. This is a collaborative study with Beam Therapeutics, Cambridge, MA under a CRADA.
The prevalent human variant G6PC1-c.247C>T/p.R83C, carrying a single G→A transition in the G6PC1 gene, is the most prevalent pathogenic mutation identified in Caucasian GSD-Ia patients and is amenable to ABE editing. We first generated a humanized G6PC1-R83C mouse strain (huR83C) by inserting the entire human G6PC1-R83C (G6PC1-c.247C→T) coding sequence into exon 1 of the mouse G6pc gene, in a manner that knocked out native mouse G6pc gene expression while placing the expression of the human G6PC1-R83C under the control of the mouse G6pc promoter/enhancer. We showed that the huR83C mice lack hepatic and renal G6Pase-α activity and manifest a human GSD-Ia phenotype.
We then evaluated the efficacy of ABE to correct the G6PC1-p.R83C variant in the huR83C mice following systemic administration of BEAM-301, the ABE editing reagent containing ABE mRNA/gRNA encapsulated in a LNP formulation. Newborn (NB) or three-week-old mice were infused with BEAM-301 and monitored for phenotypic correction up to the age of one year. We showed that the efficiency of correcting the G6PC1-R83C variant correlates with the activity of hepatic G6Pase-α restored in the edited mice. Notably, an editing efficiency of up to approximately 60% could be obtained. All edited mice harboring three or more units of hepatic G6Pase-α activity (approximately 5% editing) display an improved metabolic profile, sustain 24 hours of fasting, and survive through one year. Significantly, no decline in hepatic G6Pase-α activity was observed over the one-year study. Conversely, the untreated huR83C mice exhibit fasting hypoglycemia, with none surviving longer than to age six weeks.
An off-target analysis identified only one intronic guide-dependent off-target site, demonstrating the high precision of BEAM-301. Based on an integrated risk analysis, an edit at this off-target site is unlikely to impact the function of any associated genes, indicating that the overall risk of base editing at this off-target site is negligible, findings that led Beam Therapeutics to seek and receive an Investigational New Drug status for BEAM-301 and announce an in vivo gene-editing trial planned for early 2025 [Reference 5].
Inhibition of Wnt/β-catenin signaling reduces renal fibrosis in murine glycogen storage disease type Ia.
Renal disease is a serious long-term complication of GSD-Ia. The early kidney manifestations of GSD-Ia are impaired renal gluconeogenesis and nephromegaly caused by increased glycogen accumulation. The only therapies currently available to treat GSD-Ia are dietary therapies, which significantly alleviate metabolic abnormalities but only delay the onset of chronic kidney disease. The underlying pathological processes remain uncorrected, and glomerular hyperfiltration, hypercalciuria, hypocitraturia, and urinary albumin excretion still occur in metabolically compensated GSD-Ia patients. We previously showed that one mechanism that underlies GSD-Ia nephropathy is fibrosis mediated by activation of the renin-angiotensin system (RAS).
Wnt/β-catenin signaling regulates the expression of a variety of downstream mediators implicated in renal fibrosis, including multiple genes in the RAS. Sustained activation of Wnt/β-catenin signaling is associated with the development and progression of renal fibrotic lesions that can lead to chronic kidney disease. We examined the molecular mechanism underlying GSD-Ia nephropathy. Damage to the kidney proximal tubules is known to trigger acute kidney injury (AKI), which can, in turn, activate Wnt/β-catenin signaling. We showed that GSD-Ia mice display AKI that leads to activation of the Wnt/β-catenin/RAS axis. Renal fibrosis was demonstrated by increased renal levels of Snail1, α-smooth-muscle actin (α-SMA), and extracellular matrix proteins, including collagen-Iα1 and collagen-IV. Treating GSD-Ia mice with the CBP/β-catenin inhibitor ICG-001 significantly reduced nuclear-translocated active β-catenin and reduced renal levels of renin, Snail1, α-SMA, and collagen-IV. The results suggest that inhibition of Wnt/β-catenin signaling may be a promising therapeutic strategy for GSD-Ia nephropathy [Reference 4].
Additional Funding
- CRISPR Therapeutics (Cambridge, MA), under a cooperative research and development agreement (CRADA)
- Beam Therapeutics, Inc (Cambridge, MA), under a CRADA
Publications
- Molecular mechanism underlying hepatic autophagy impairment in glycogen storage disease type Ib. Hum Mol Genet 2023 32:262–275
- Gene therapy and genome editing for type I glycogen storage diseases. Frontiers Mol Med 2023 3:1167091
- CRISPR/Cas9-based double strand oligonucleotide insertion strategy corrects metabolic abnormalities in murine glycogen storage disease type Ia. J Inherit Metab Dis 2023 46(6):1147–1158
- Inhibition of Wnt/β-catenin signaling reduces renal fibrosis in murine glycogen storage disease type Ia. Biochim Biophys Acta Mol Basis Dis 2023 1870(1):166874
- Base-editing corrects metabolic abnormalities in a humanized mouse model for glycogen storage disease type-Ia. Nat Commun 2024 15(1):9729
Collaborators
- Troy Carlo, PhD, Prime Medicine Inc, Cambridge, MA
- Alessandra Eva, PhD, Istituto Giannina Gaslini, Genoa, Italy
- Christopher Hart, PhD, Prime Medicine Inc, Cambridge, MA
- Michael Kahn, PhD, Beckmann Research Institute, City of Hope, Duarte, CA
- Matthew F. Starost, PhD, Diagnostic & Research Services Branch, Division of Veterinary Resources, NIH, Bethesda, MD
Contact
For more information, email chouja@mail.nih.gov or visit https://www.nichd.nih.gov/research/atNICHD/Investigators/chou.