
Chromatin Remodeling and Gene Activation

- David J. Clark,
PhD, Head, Section on Chromatin and Gene Expression - Peter Eriksson, PhD, Staff Scientist
- Hemant K. Prajapati, PhD, Research Fellow
- Lalita Panigrahi, PhD, Visiting Fellow
- Zhuwei Xu, PhD, Visiting Fellow
- Kenneth Wu, PhD, Postdoctoral Intramural Research Training Award Fellow
Aberrant gene regulation is the basis of many disease states. Our main objective is to understand how genes are activated for transcription in the context of chromatin structure. Chromatin is not merely a packaging system for DNA in eukaryotic cells; it also participates in gene regulation. It is thought that gene regulation involves either attenuation of the inherently repressive properties of nucleosomes to facilitate gene expression, or enhancement of those properties to ensure complete repression. Such events are choreographed by DNA sequence–specific transcription factors (activators and repressors) and chromatin remodeling complexes. The latter can be divided into two groups: histone- or DNA–modifying enzymes that implement the "epigenetic code", and ATP–dependent remodeling machines that move or displace nucleosomes. Our studies are focused primarily on the roles of the ATP–dependent chromatin remodelers in gene regulation.
The packaging of chromosomal DNA in eukaryotic nuclei occurs at several structural levels (Figure 1). The basic structure resembles beads-on-a-string, in which the beads are nucleosome cores and the string is connecting linker DNA. The nucleosome core is composed of about 147 bp of DNA wrapped in 1.7 negative superhelical turns around a central histone octamer containing two molecules each of the four core histones (H2A, H2B, H3, and H4). The nucleosomal filament coils spontaneously into a heterogeneous higher-order structure that is about 30 nm wide, facilitated by linker histones, and is organized into loop domains by other chromatin proteins, such as CTCF and condensin. Inactive chromatin (heterochromatin) is generally much more condensed than active chromatin (euchromatin) and is associated with specific proteins such as heterochromatin protein 1 (HP1) and Polycomb repressor complexes.
Figure 1. DNA packaging in the nucleus: to what extent does chromatin compaction limit access to the DNA?
DNA is packaged into the nucleus by histones. The basic structural subunit of chromatin is the nucleosome core, which contains about 147 bp of DNA wrapped nearly twice around a central octamer of core histones. Nucleosomes are regularly spaced along the DNA like beads on a string; the intervening DNA is called the linker DNA and is bound by linker histone (H1). The beads-on-a-string fiber spontaneously condenses into a heterogeneous fiber of about 30 nm width. Genomic regions rich in repetitive elements form constitutive heterochromatin in all cells, in which the chromatin fiber is even more condensed. Facultative heterochromatin is formed on genes that should be permanently silent in a specific differentiated cell type. Heterochromatin is densely packed and darkly staining in the electron micrograph shown here. Euchromatin is less condensed (light staining) and contains active genes. We are interested in determining to what extent chromatin limits DNA accessibility. Figure adapted from Chereji et al. Genome Res 2019;29:1985-1995.
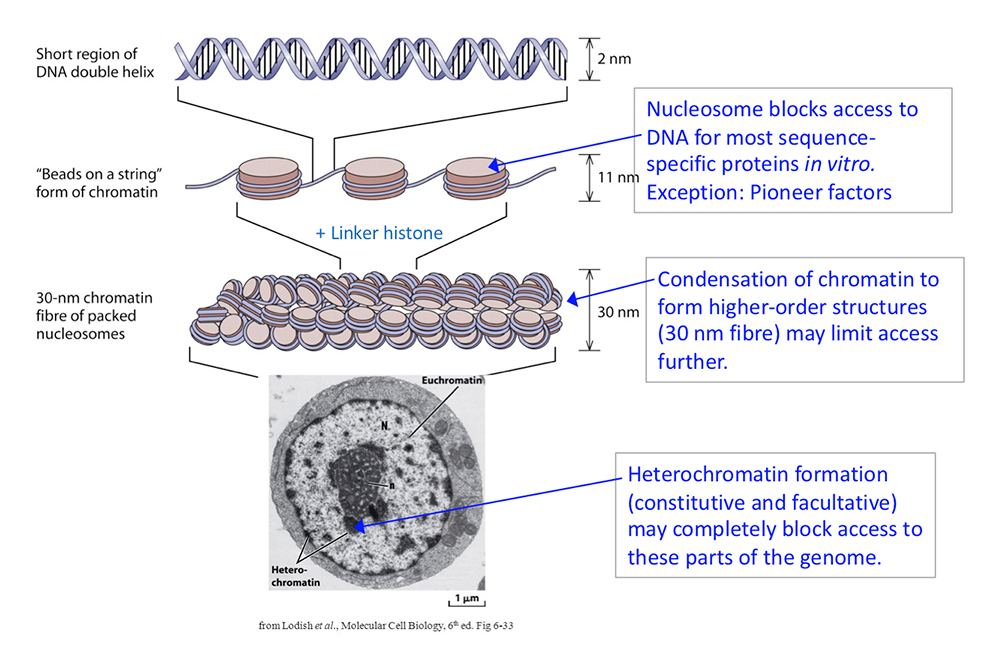
Figure 1. DNA packaging in the nucleus: to what extent does chromatin compaction limit access to the DNA?
DNA is packaged into the nucleus by histones. The basic structural subunit of chromatin is the nucleosome core, which contains about 147 bp of DNA wrapped nearly twice around a central octamer of core histones. Nucleosomes are regularly spaced along the DNA like beads on a string; the intervening DNA is called the linker DNA and is bound by linker histone (H1). The beads-on-a-string fiber spontaneously condenses into a heterogeneous fiber of about 30 nm width. Genomic regions rich in repetitive elements form constitutive heterochromatin in all cells, in which the chromatin fiber is even more condensed. Facultative heterochromatin is formed on genes that should be permanently silent in a specific differentiated cell type. Heterochromatin is densely packed and darkly staining in the electron micrograph shown here. Euchromatin is less condensed (light staining) and contains active genes. We are interested in determining to what extent chromatin limits DNA accessibility. Figure adapted from Chereji et al. Genome Res 2019;29:1985-1995.
Chromatin structure apparently presents a formidable obstacle to sequence-specific transcription factors searching for their specific binding sites. Most of these factors bind very weakly, if at all, to a cognate site within a nucleosome, unless the site is situated just inside the nucleosome, where the histone–DNA contacts are weakest. However, there are some transcription factors, termed "pioneer" factors, that have evolved to bind nucleosomal sites with high affinity. Many pioneer factors are critical for development, given that they have the potential to invade chromatin.
In vitro studies demonstrating that nucleosomes inhibit transcription factor binding resulted in the discovery of the ATP–dependent chromatin remodeling enzymes, which facilitate access to the DNA. These enzymes can slide nucleosomes along DNA, move the histone octamer from one DNA molecule to another, and drive nucleosomal conformational changes. Examples include the yeast SWI/SNF, RSC, ISW1, and CHD1 multi-subunit complexes, and their metazoan counterparts, including human BAF and PBAF, SNF2H/L, and CHD complexes. These enzymes play a critical role in gene regulation. They act at promoters and other regulatory elements to facilitate nucleosome removal, allowing transcription factors to bind and subsequent recruitment of RNA polymerase II. Mutations in genes encoding chromatin remodeler subunits are associated with autism spectrum disorder and various human diseases, including many cancers (e.g., pediatric malignant rhabdoid tumors).
Measuring genome accessibility in living cells
The general view is that nucleosomes and chromatin structure play a central role in gene regulation by restricting access to genomic DNA. Experiments measuring DNA accessibility in isolated nuclei (e.g., by MNase–Seq, ATAC–Seq, qDA–Seq) have established that nucleosomes block access to DNA. Studies from many groups have confirmed this observation, and is stated as a fact in the introductions to most chromatin papers (including our own) and in reviews of the chromatin field. Indeed, the observation that nucleosomes block access to DNA is central to current models of gene regulation, as mentioned above. However, we have now established that isolated nuclei do not represent an accurate model for living cells, because chromatin dynamics are "frozen" when cells are disrupted. Disruption results in the loss of metabolites, including the ATP required for remodeling.
Previously, we compared the accessibility of the genome in isolated nuclei and in living yeast cells using DNA methylases. Remarkably, we found that the yeast genome is globally accessible in living cells, unlike in nuclei. That is, nucleosomes do not block access to genomic DNA in vivo. Briefly, we showed that methylation by the Dam DNA methylase (which methylates A in the sequence GATC) is blocked by nucleosomes in isolated nuclei. However, expression of Dam in living cells using an inducible promoter results in methylation of the entire genome, with minimal interference from nucleosomes. Using a different DNA methylase, M.SssI (which methylates C in CG), together with nanopore long-read sequencing, we showed that centromeric nucleosomes, unlike canonical nucleosomes, are exceptionally stable, protecting their DNA from methylation in vivo. The silenced mating type loci are also much less accessible than the rest of the genome. We showed that at least three ATP–dependent chromatin remodelers (RSC, ISW1, and CHD1) contribute to nucleosome dynamics in vivo, using a degron approach to detect nucleosome movements in living cells as remodelers are depleted.
Our data demonstrate that nucleosomes are in a continuous state of flux in living cells, but static in nuclei, presumably because of the loss of critical factors during isolation. This flux may involve nucleosome sliding, nucleosome removal and replacement, and/or nucleosome conformational changes, catalyzed by ATP–dependent chromatin remodelers (Figure 2). We propose that the various remodelers compete with one another in vivo, continually moving nucleosomes to different positions, resulting in a nucleosome flux that renders the yeast genome essentially transparent to transcription factors and other DNA–binding proteins.
Figure 2. Chromatin flux model to explain the difference in genome accessibility in nuclei and living cells
We propose that nucleosomes are stationary in nuclei, protecting the DNA they contain (top panel). The Dam DNA methyltransferase only methylates the short stretches of linker DNA between regularly spaced nucleosomes. However, in living cells, Dam methylates virtually the entire genome (bottom panel), because the nucleosomes are in flux. Nucleosomes may be continuously removed and replaced, or shunted from side to side, or opened up, as shown, resulting in DNA exposure and methylation. We propose that the flux is generated by the competing activities of the various ATP–dependent chromatin remodelers (such as RSC, SWI/SNF, ISW1, and CHD1). Adapted from Reference 3.
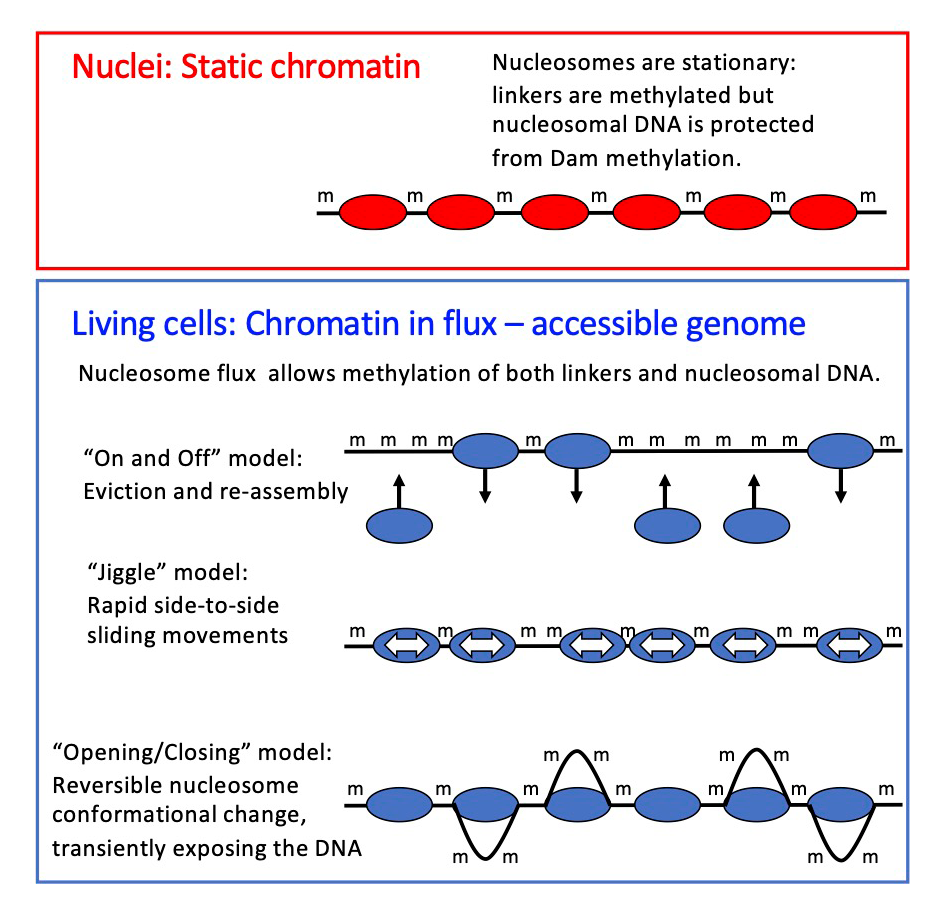
Figure 2. Chromatin flux model to explain the difference in genome accessibility in nuclei and living cells
We propose that nucleosomes are stationary in nuclei, protecting the DNA they contain (top panel). The Dam DNA methyltransferase only methylates the short stretches of linker DNA between regularly spaced nucleosomes. However, in living cells, Dam methylates virtually the entire genome (bottom panel), because the nucleosomes are in flux. Nucleosomes may be continuously removed and replaced, or shunted from side to side, or opened up, as shown, resulting in DNA exposure and methylation. We propose that the flux is generated by the competing activities of the various ATP–dependent chromatin remodelers (such as RSC, SWI/SNF, ISW1, and CHD1). Adapted from Reference 3.
However, budding yeast does not have the heterochromatin typical of the cells of higher organisms. Heterochromatin is generally associated with a high level of chromatin condensation and gene repression and is expected to be much less accessible than transcriptionally active euchromatin. To address this important question, we extended our genome accessibility studies to human cell lines, specifically MCF7 breast cancer cells and MCF10A cells [Reference 1]. We observed that heterochromatin is methylated only slightly more slowly than euchromatin; that is, heterochromatin is also generally accessible in living cells. Again, we propose that ATP–dependent remodelers are responsible for maintaining DNA accessibility in live cells, and we invoke the same models for nucleosome dynamics that we proposed for yeast cells (Figure 2). Our observations have profound implications for the chromatin field, requiring a re-examination of the roles of the chromatin remodelers in gene regulation, and of the extent to which packaging the genome into nucleosomes is actually repressive.
We continued our investigation of the roles of ATP–dependent chromatin remodelers in maintaining nucleosome dynamics in living yeast cells by focusing on the ISW1 and CHD1 nucleosome-spacing enzymes [Reference 2]. We found that ISW1 and CHD1 together suppress nucleosome dynamics in living cells, that is, the genome is methylated faster in the absence of both remodeling enzymes. We propose that ISW1 and CHD1 slide nucleosomes to create an ordered spaced nucleosome array (i.e., the 'beads on a string') and to suppress histone exchange (the cycle of histone removal from DNA and their replacement), resulting in reduced nucleosome dynamics and therefore in slower DNA methylation. In the absence of ISW1 and CHD1, their nucleosome-sliding activities are lost but histone exchange increases, perhaps attributable to a nucleosome flux–creating remodeler, resulting in an overall increase in dynamics and methylation rate. Our working model is shown in Figure 3. Currently, we are testing various aspects of our nucleosome dynamics model in yeast and in human cell lines.
Figure 3. Working model for dynamic chromatin in vivo [from Reference 2].
Remodeled nucleosomes may be displaced, slid along the DNA, and/or conformationally altered. Genes cycle between a highly dynamic remodeled state (bottom) and a less dynamic ordered state (top), attributable to opposing remodeling activities. RSC, SWI/SNF, and/or INO80 may be responsible for creating dynamic remodeled chromatin, with transcription playing a minor role. ISW1 and CHD1 reduce nucleosome dynamics by suppressing remodeling events that may result in histone exchange (e.g., nucleosome displacement or destabilized, conformationally altered nucleosomes). In the absence of Isw1 and Chd1, chromatin persists in the highly dynamic remodeled state, because of the unopposed activities of the flux-creating remodelers. The promoter is shown occupied by a transcription complex, which cycles on and off the DNA in vivo (blue box).
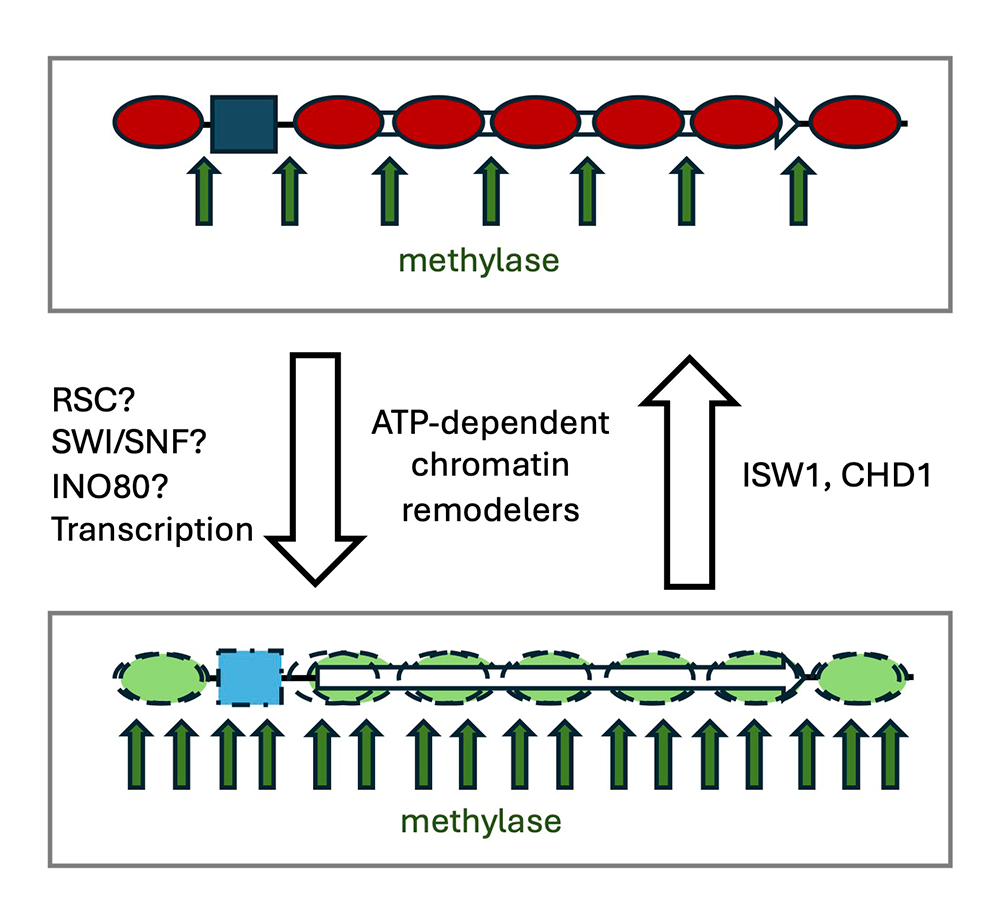
Figure 3. Working model for dynamic chromatin in vivo [from Reference 2].
Remodeled nucleosomes may be displaced, slid along the DNA, and/or conformationally altered. Genes cycle between a highly dynamic remodeled state (bottom) and a less dynamic ordered state (top), attributable to opposing remodeling activities. RSC, SWI/SNF, and/or INO80 may be responsible for creating dynamic remodeled chromatin, with transcription playing a minor role. ISW1 and CHD1 reduce nucleosome dynamics by suppressing remodeling events that may result in histone exchange (e.g., nucleosome displacement or destabilized, conformationally altered nucleosomes). In the absence of Isw1 and Chd1, chromatin persists in the highly dynamic remodeled state, because of the unopposed activities of the flux-creating remodelers. The promoter is shown occupied by a transcription complex, which cycles on and off the DNA in vivo (blue box).
Publications
- Nucleosome dynamics render heterochromatin accessible in living human cells. Nat Commun 2025 16:4577
- The ISW1 and CHD1 chromatin remodelers suppress global nucleosome dynamics in living yeast cells. Sci Adv 2025 11:eadw7108
- The yeast genome is globally accessible in living cells. Nat Struct Biol 2025 32(2):247-256
Collaborators
- Hussam Alkaissi, MD, MS, NIDDK, Bethesda, MD
- David Levens, MD, PhD, Laboratory of Pathology, Center for Cancer Research, NCI, Bethesda, MD
- Karel Pacak, MD, PhD, DSc, Section on Medical Neuroendocrinology, NICHD, Bethesda, MD
Contact
For more information, email chitnisa@mail.nih.gov or visit https://www.nichd.nih.gov/research/atNICHD/Investigators/chitnis.