
Physiological, Biochemical, and Molecular-Genetic Events Governing the Recognition and Resolution of RNA/DNA Hybrids

- Robert J. Crouch,
PhD, Head, Section on Formation of RNA - Susana M. Cerritelli, PhD, Staff Scientist
- Kiran Sakhuja, MS, MSc, Biologist
Ribonucleases H (RNases H) are considered essential enzymes in multicellular organisms, thereby placing the genes that encode the enzymes in the housekeeping category. Damaged DNA is a leading cause of many human diseases and disorders. We study the formation and resolution of RNA/DNA hybrids, which occur during DNA replication and RNA transcription. Such hybrid molecules may lead to increased DNA damage, but may also play critical roles in normal cellular processes. We are interested in how RNA/DNA hybrids are resolved and in the role that RNases H play in their elimination. Two classes of RNases H, Class I and Class II, are present in most organisms.
Patients with mutations in the RNASEH1 gene exhibit typical mitochondrial myopathy symptoms (neuromuscular disorder). We were the first to show that RNase H1 is essential for the maintenance of mitochondrial DNA. More than 1,000 proteins are targeted to mitochondria, many of which, when mutated, are known causes of mitochondrial myopathies. Mice deleted for the Rnaseh1 gene arrest embryonic development at day 8.5 because of failure to amplify mitochondrial DNA.
Aicardi-Goutières syndrome (AGS), a severe neurological disorder with symptoms appearing at or soon after birth, can be caused by defective human RNase H2. As many as 38 Mendelian genotypes may result in a type I interferonopathy, including mutations in each of the genes encoding the subunits of the heterotrimeric RNase H2, the hallmark of which is activation of the innate immune response.
Figure 1. Substrates for RNases H
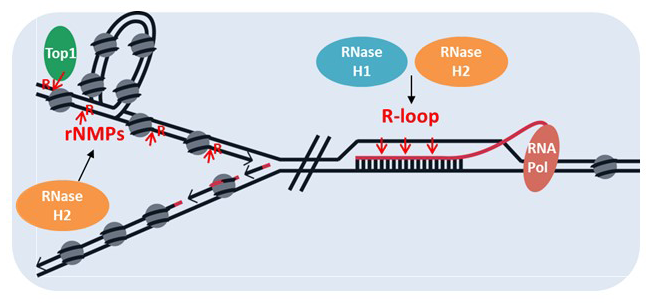
Figure 1. Substrates for RNases H
RNA/DNA hybrids, ribonucleotides embedded in DNA: overlapping and disparate resolution by RNases H and the RNA exosome
The catalytic mechanisms of RNase H1 and RNaseH2 are very similar, but their overall structures are very different. In vitro, both types hydrolyze the RNA of RNA/DNA hybrids. However, RNase H1 requires a minimum length of four rNMPs in an RNA/DNA hybrid, whereas RNase H2 recognizes and incises at a single rNMP embedded in DNA. Moreover, each enzyme has a unique dominant function leading to different symptoms in patients with RNase H deficiencies: RNase H1 defects lead to mitochondrial myopathy in mice and human patients, whereas RNase H2 mutations lead to a type I interferonopathy. Interestingly, our mouse model with one of the mutations found in patients (G37S-RNASEH2A), which in humans causes interferonopathy, results in perinatal lethality in mice.
Separation of function mutations for RNase H1
Our results obtained in the past indicate that the biochemical properties of mice and human RNases H1 are quite similar. Both enzymes have two isoforms, translated from a single mRNA, that produce mitochondrial and nuclear forms. We found that absence of RNase H1 during development caused defects in mitochondrial DNA replication, resulting in embryonic arrests and, possibly, obscuring any role of the nuclear RNase H1. In 2025, we generated a mouse model that expresses only the mitochondrial RNase H1 in the hope of uncovering functions of the more abundant nuclear RNase H1. The mice are viable and fertile, suggesting that RNase H1 has no essential function in the nucleus, at least under physiological conditions.
Separation of function mutations for RNase H2
RNase H2 has two activities: hydrolysis of RNA in RNA/DNA hybrids and incision of single ribonucleotides (rNMPs) embedded in DNA, otherwise known as ribonucleotide excision repair (RER). We generated a mutant of RNase H2 that retained the ability to hydrolyze RNA in RNA/DNA hybrids but failed to incise at rNMPs in DNA (ribonucleotide excision defective: RED). Mice homozygous for the RED mutation were embryonic-lethal at E9–E10, the same as RNase H2–null mice. We also generated a mutation that retained significant RER activity but less than a few percent of hybrid activity (hybrid defective: HD). HD mice are viable and fertile.
Double mutant of RNase H1 and RNase H2 defective for R-loop processing mice are viable and fertile.
The simple conclusion from these studies is that RNase H1 is needed for mitochondrial DNA replication but has little or no nuclear function, whereas the major role of RNase H2 is removal of single rNMPs in DNA.
We crossed homozygous mice that only express mitochondrial RNase H1 form with homozygous mice that have RNase H2 defective in processing RNA/DNA hybrids. These mice are viable. We are studying the R-loop distribution in these mutants to determine the role of RNase H1 and RNase H2 in R-loop resolution.
The RNA exosome and RNases H cooperate to suppress R-loop–mediated genome instability.
In eukaryotes, the RNA exosome is a major 3′-5′ RNA degradation, multi-subunit complex, which degrades cryptic and defective transcripts, preventing their engagement with DNA and the formation of R-loops. In collaboration with Aziz El Hage and David Tollervey, we are studying the interactions between the RNA exosome and RNases H in suppressing R-loop–mediated genome instability.
Using the yeast S. cerevisiae, we found that cells defective in both RNase H and RNA exosome activities are hypersensitive to the drug hydroxyurea (HU), which induces replicative stress by depleting the cellular dNTP supply. RNase H2–RED variant and over-expression of RNase H1 partially suppressed the growth defects in presence of HU, suggesting that some of the problems in these cells are caused by harmful R-loops.
We determined that R-loop–mediated genome instability and hyper-recombination are increased in mutants defective for RNase H and the RNA exosome, concluding that both pathways converge to prevent R-loop accumulation. We are in the process of identifying the RNA/DNA hybrids that are presents in these cells.
Publications
- Two RNase H2 mutants with differential rNMP processing activity reveal a threshold of ribonucleotide tolerance in DNA for embryonic development. Cell Rep 2018 25:1135–1145
- High density of unrepaired genomic ribonucleotides leads to Topoisomerase 1-mediated severe growth defects in absence of ribonucleotide reductase. Nucleic Acids Res 2020 48:4274–4297
- RNA abasic sites in yeast and human cells. Proc Natl Acad Sci USA 2020 117:20689–20695
- A common transcriptional mechanism involving R-loop and RNA abasic site regulates an enhancer RNA of APOE. Nucleic Acids Res 2022 50(21):12497–12514
Collaborators
- Charles Bou-Nader, PhD, Emory University, Atlanta, GA
- Frederic Chedin, PhD, University of California Davis, Davis, CA
- Louis Dye, BS, Microscopy and Imaging Core, NICHD, Bethesda, MD
- Aziz El Hage, PhD, University of Edinburgh, Edinburgh, United Kingdom
- Sergei Gaidamakov, PhD, Section on Molecular and Cellular Biology, NICHD, Bethesda, MD
- Alexander Grinberg, DVM, NICHD, Bethesda, MD
- James Iben, PhD, Molecular Genomics Core, NICHD, Bethesda, MD
- David Tollervey, PhD, University of Edinburgh, Edinburgh, United Kingdom
- Jinwei Zhang, PhD, Laboratory of Molecular Biology, NIDDK, Bethesda, MD
Contact
For more information, email crouchr@mail.nih.gov or visit https://www.nichd.nih.gov/research/atNICHD/Investigators/crouch.