
Molecules and Therapies for Craniofacial and Dental Disorders
- Rena D’Souza,
MS, DDS, PhD, Head, Section on Craniofacial Genetic Disorders, and Director of the National Institute of Dental and Craniofacial Research - Masae Yamada, DDS, PhD, Staff Scientist
- Aye Chan Myo, DDS, PhD, Postdoctoral Fellow
- Jeremie Oliver Piña, PhD, MS, MBA, Postdoctoral Fellow
- Resmi Raju, PhD, Postdoctoral Fellow
- Parna Chatteraj, MS, Lab Manager
Embryonic development of the craniofacial complex requires tightly controlled molecular crosstalk between proliferating and differentiating cell networks to form the most intricate structures in the human body. During embryogenesis, perturbations in the environmental and/or genetic milieu can negatively affect craniofacial development. While considerable progress has been made in studying isolated genetic mutations leading to syndromic and non-syndromic craniofacial disorders, the broad molecular-genetic mechanisms driving morphogenetic events have yet to be sufficiently explored and understood. Such gaps in our understanding have restricted clinical treatment options for patients affected by common developmental anomalies, such as cleft palate or tooth agenesis. Thus, there is a strong biologic rationale for a thorough investigation of basic molecular mechanisms driving craniofacial structure morphogenesis, which may pave the way toward translatable therapeutic developments for patients.
The overarching goal of our research program is to conduct basic and translational studies of genetic and molecular mechanisms involved in craniofacial development, with the primary aim of unveiling novel regulatory molecules and putative patient-centric therapeutic solutions for craniofacial and dental disorders. Among the molecular pathways known to control craniofacial development is Wnt/β-catenin signaling. Wnts are also well known as upstream effectors of osteogenesis and odontogenesis. Our lab demonstrated, for the first time, the successful in utero correction of cleft palate defects in a Pax9–/– mouse genetic model by small-molecule neutralizing therapy targeting Wnt–antagonizing proteins. We are now actively investigating additional drug-delivery approaches and molecular targets for the modulation of Wnt signaling in vivo to permit targeted correction of both cleft palate and tooth agenesis. Our research group employs basic principles of developmental biology, next-generation sequencing, regenerative medicine, tissue engineering, and drug delivery models to identify and validate novel approaches to restore molecular equilibrium in genetic models of the highly relevant human diseases cleft palate and tooth agenesis. Actively fostering collaborations with both intramural and extramural investigators, from basic scientists, engineers, to clinicians, the lab aims to pioneer innovative approaches toward the treatment of patients affected by craniofacial disorders of development. Through these rigorous and robust preclinical, proof-of-principle studies, we hope to open up pathways toward novel clinical trials for the treatment of previously unpreventable disorders affecting the craniofacial complex.
Figure 1. Wnt–mediated osteogenesis defines palatal shelf fusion.
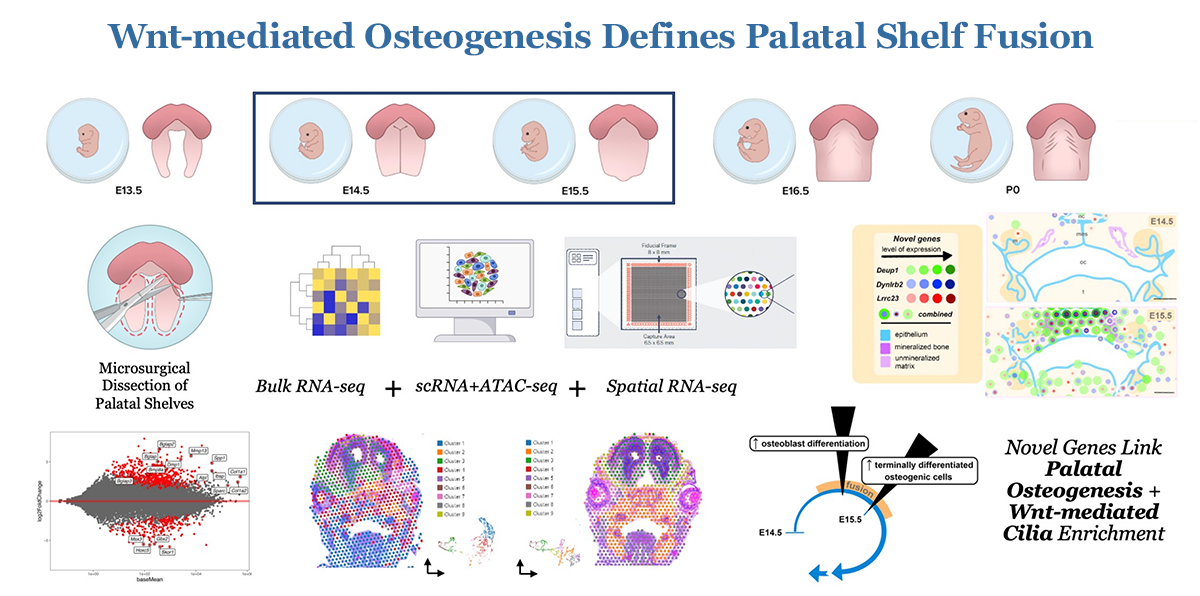
Figure 1. Wnt–mediated osteogenesis defines palatal shelf fusion.
Toward a novel Wnt–driven therapeutic approach for cleft palate
Cleft palate (CP), together with cleft lip, is among the most common birth defects in humans, occurring in up to 1 in 500 live births. Such birth defects can inflict heavy physical, mental, psychosocial, and financial burdens on patients and their caregivers throughout life, often requiring many stages of complex surgical correction with varying success rates. Hence, there remains a substantial need for innovative approaches to alleviate the burden of postnatal care for these patients. While considerable progress has been made in studying isolated genetic mutations leading to syndromic and non-syndromic cleft disorders, the broad molecular-genetic mechanisms driving osteogenic differentiation from palatal shelf out-growth, elevation, and fusion events have yet to be sufficiently explored and understood. Such gaps in our understanding have restricted clinical treatment options for patients affected by cleft disorders.
Figure 2. Pax9 orchestrates Wnt signaling pathway genes in all cell types.
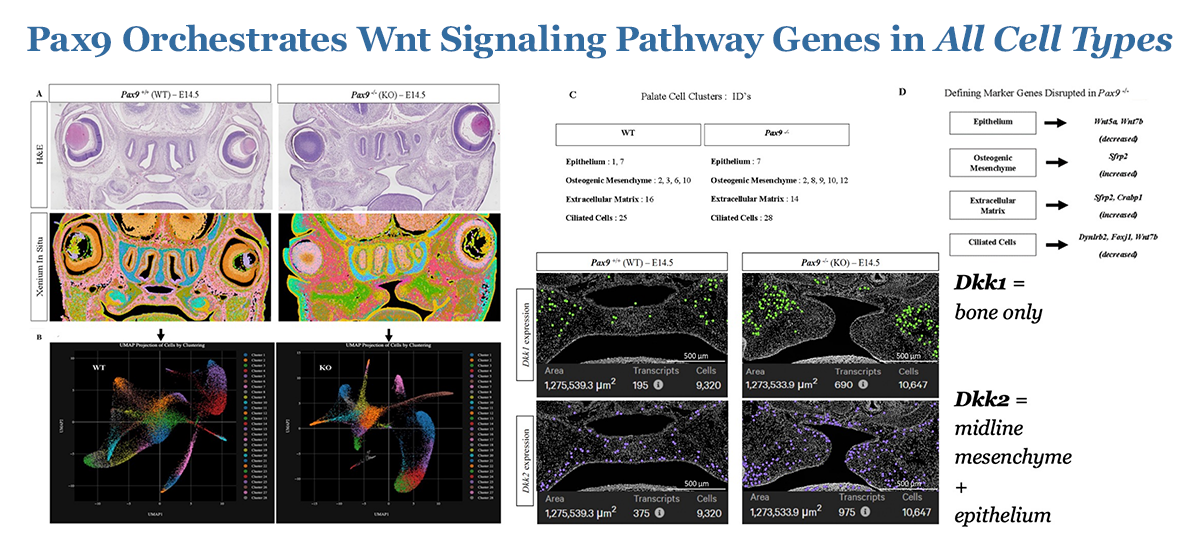
Figure 2. Pax9 orchestrates Wnt signaling pathway genes in all cell types.
The objective of our work is to investigate the molecular-genetic mechanisms driving palatogenesis to better understand the unique signaling milieu requisite for proper palate formation. If properly explored, such information can be applied to the development of novel therapeutics that can benefit individuals with isolated or syndromic CP defects, who face complex surgeries and the arduous burden of life-long care. The long-term goal of this research is to investigate the spatio-temporal molecular mechanisms driving osteogenic differentiation in normal palate development and in CP dysmorphogenesis, using Pax9–/– as a model. Differential multi-omic profiles of expression in spatial biological context are now beginning to unveil the molecular framework for the development of novel therapeutic strategies to optimize signaling environments in the palate. We are testing the following hypotheses: (1) Wnt signaling effector function is critical for osteogenesis of the embryonic palate; and (2) loss of the up-stream master regulator of Wnt signaling homeostasis, Pax9, results in the disruption of palatal osteogenesis via perturbed formation of extracellular matrix components critical for the palate to fuse. Over the last several years, we have made significant progress in defining the spatio-temporal transcriptomic profile of embryonic palate osteogenesis using unbiased stage-specific signature mapping of cell populations in the normal murine secondary palate. We then extended these studies to differentially analyze the multi-omic profile of the Pax9 knockout (KO) murine cleft palate during many phases of development, highlighting the likely pathologic role of key Wnt signaling modulators in disrupting normal development of the palate. Now, we look toward developing and piloting novel therapeutic strategies to target these molecules of interest identified in our prior work in emerging pre-clinical and subsequent clinic studies, specifically for potential application in alveolar cleft treatment, which is severely lacking. The validation of a translational palate-specific therapeutic system would be a meaningful step toward bettering future clinical outcomes for a very vulnerable patient population worldwide.
Figure 3. Wnt signaling drives palate osteogenesis and fusion.
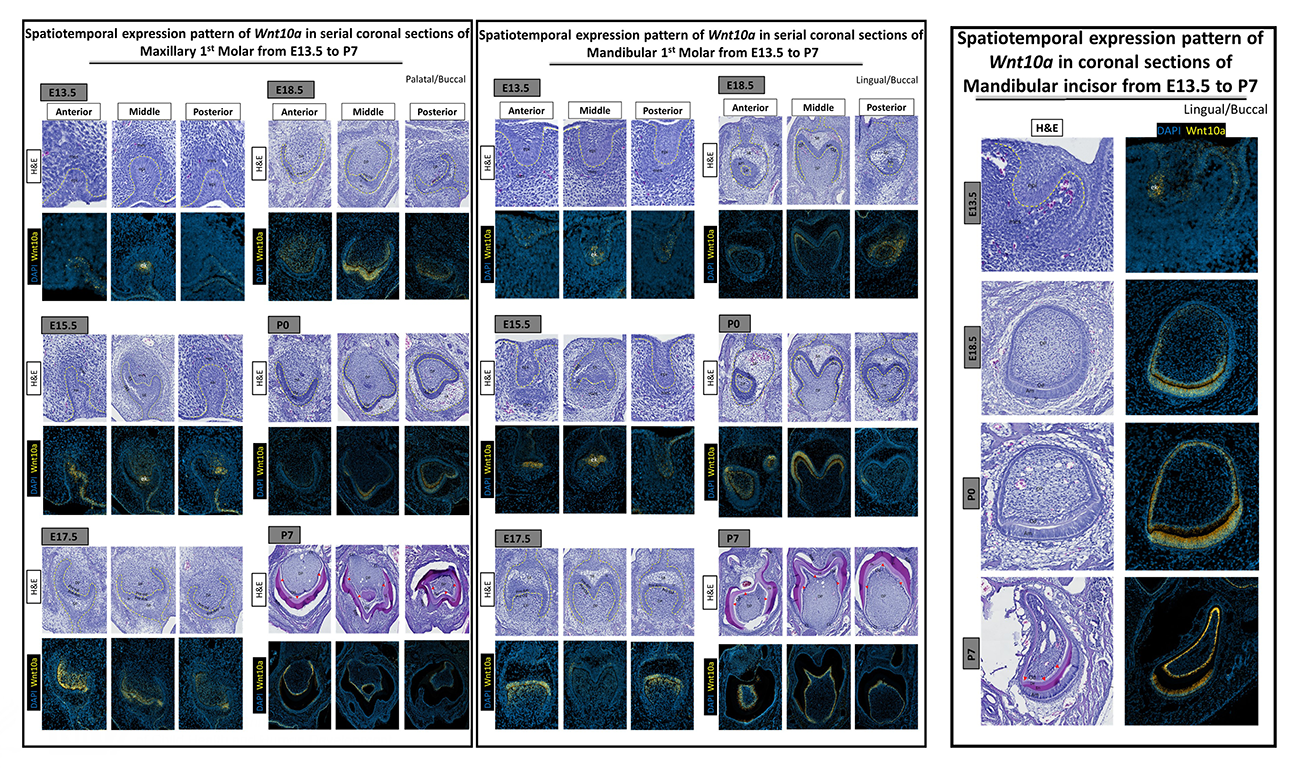
Figure 3. Wnt signaling drives palate osteogenesis and fusion.
Profiles of Wnt pathway gene expression during tooth morphogenesis
Mouse and human genetic studies indicate key roles of the Wnt10a ligand in odontogenesis. Despite the advances, little is known about the temporo-spatial modulation of Wnt10a and whether its expression profiles explain the reciprocal epithelial-mesenchymal signaling events that drive morphogenesis and cell differentiation. We systematically compared the profiles of Wnt10a in developing murine molars and incisors with the Wnt signaling pathway inhibitors Sost and Dkk1, using multiplex in situ hybridization and single-cell RNA-sequencing (scRNA-seq). During tooth bud morphogenesis, Wnt10a transcripts were restricted to the epithelium, in contrast to the localization of Sost and Dkk1 to the dental mesenchyme. At E15.5, there was a marked shift of Wnt10a from dental epithelium to mesenchyme, while Sost and Dkk1 expression remained enriched in the mesenchyme. By E18.5 and P0 (postnatal day 0), Wnt10a expression coincided with the gradients of ameloblast and odontoblast differentiation from cusp to apical regions. Interestingly, Sost and Dkk1 co-expressed with Wnt10a in odontoblasts at these stages. At P7 and 14, following dentin and enamel mineralization, Wnt10a was confined to odontoblasts, while Wnt modulators were reduced or absent in the molars, but intense signals were continuously present in ameloblasts (Wnt10a) and odontoblasts (Wnt10a, Sost, and Dkk1) towards the proximal end of incisors near the cervical loop. scRNA-seq confirmed in situ expression signatures of target genes to clusters containing dental cells. These data provide cell type–specific insight into the role of Wnt signaling mediators during epithelial-mesenchymal interactions in odontogenesis. Our results provide a framework for developing well timed therapies that target Wnt signaling for the reversal of tooth agenesis and for regenerating the dentin-pulp complex after injury.
Figure 4. Wnt10a and Wnt modulators
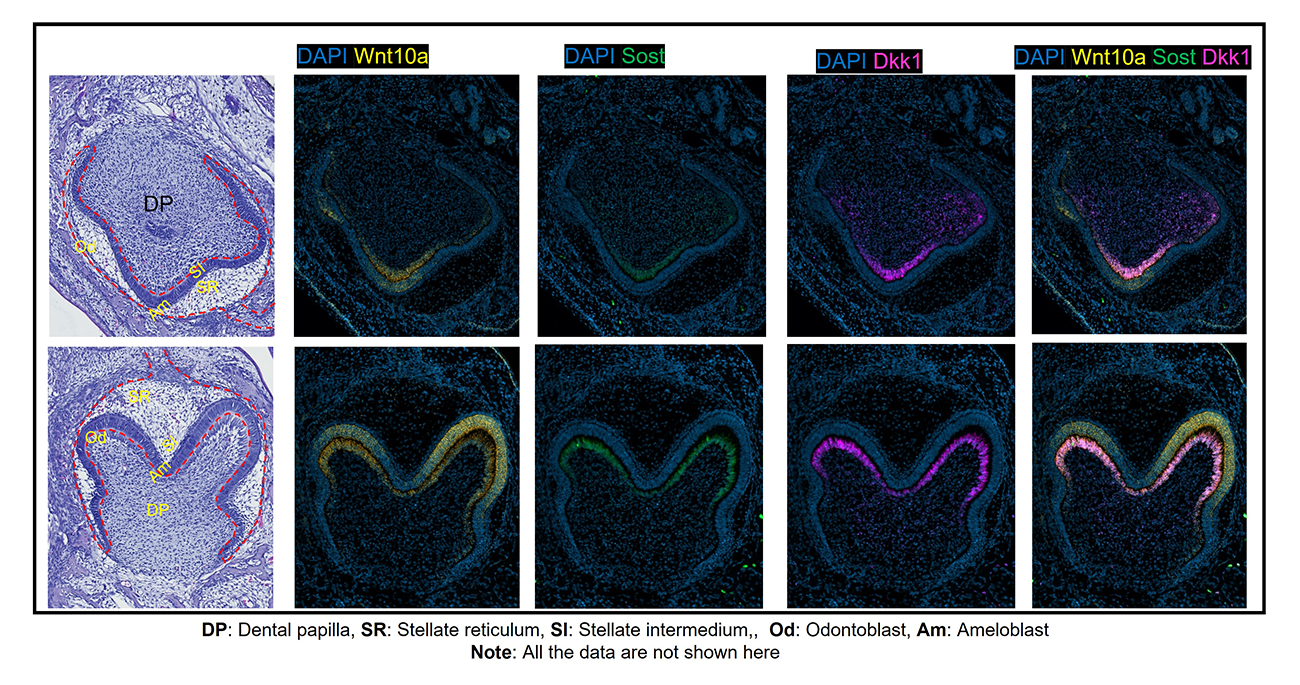
Figure 4. Wnt10a and Wnt modulators
In vivo molecular consequences of Sosdc1 deficiency on Pax9– and/or Msx1–dependent signaling events that control maxillary vs. mandibular incisors
Tooth agenesis is a developmental anomaly defined by the lack of one or more teeth (excluding third molars) resulting from to early failures in tooth formation. Several genes potentially causing tooth agenesis have been studied. A familial autosomal dominant hypodontia (tooth agenesis) was demonstrated to be caused by a point mutation in the MSX1 and PAX9 genes. However, how MSX1 and PAX9 control the patterning of tooth agenesis requires further investigation. Interestingly, Pax9+/–/Msx1+/– mutant mice exhibit selective oligodontia (missing lower incisors only) and offer an excellent opportunity to study the Pax9– and Msx1–dependent tooth agenesis patterning. Concurrently, we and others have shown that the transcription factors Msx1 and Pax9 modulate the functions of Bmp and Wnt during odontogenesis in a mouse model. Interestingly, we found that transgenic inhibition of a known bifunctional BMP and Wnt antagonist, Sosdc1, was able to rescue the lower incisor in Pax9– and Msx1–deficient mice (Pax9+/–; Msx1+/–/Sosdc1–/–). However, how exclusively Msx1 and/or Pax9 interact with Sosdc1 in the mandibular incisor’s domain vs. the maxillary incisor’s domain requires further investigation. Therefore, our ongoing experiments (bulk RNA, multiplex in situ hybridization, and multi-omics analysis) will explore the molecular consequences of Sosdc1 deficiency on Pax9– and/or Msx1–dependent signaling events in the maxillary vs. mandibular incisors domain.
Figure 5. In vivo molecular consequences of Sosdc1 deficiency on Pax9– and/or Msx1–dependent signaling events that control maxillary vs mandibular incisors
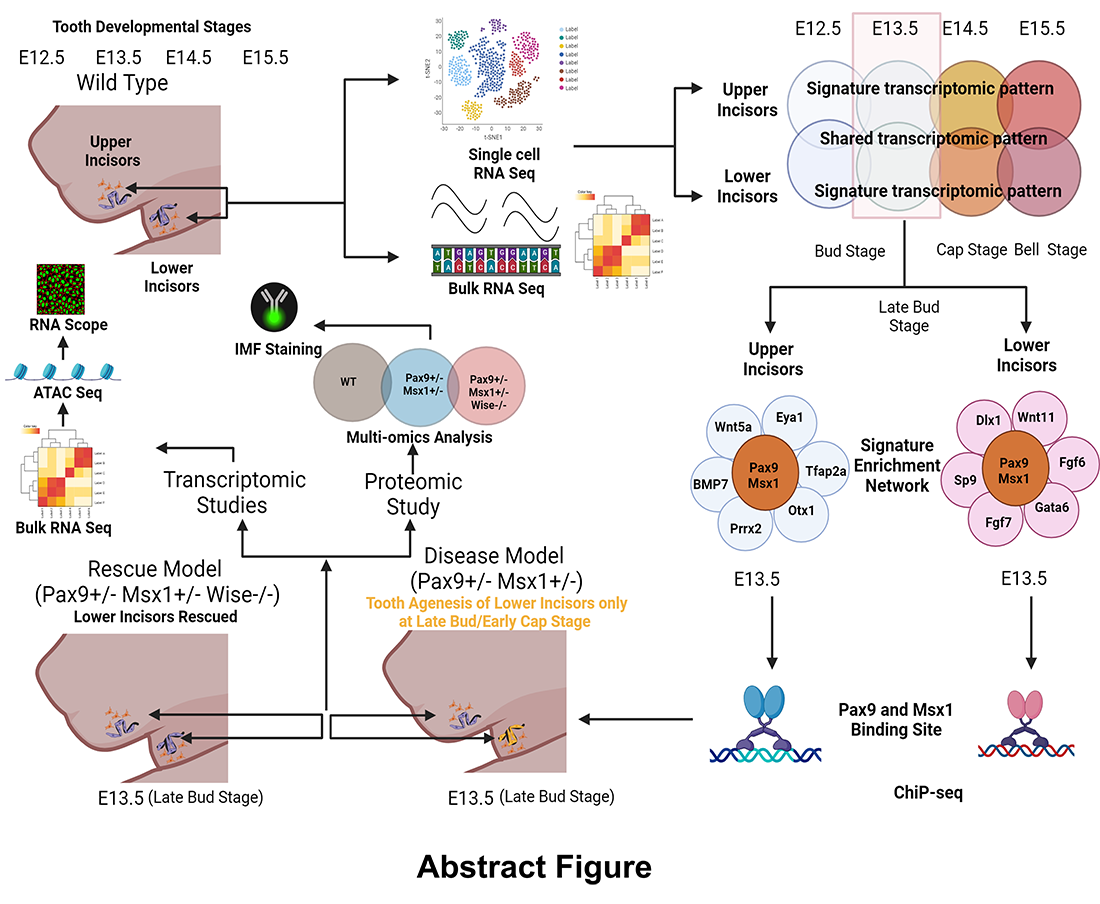
Figure 5. In vivo molecular consequences of Sosdc1 deficiency on Pax9– and/or Msx1–dependent signaling events that control maxillary vs mandibular incisors
Exploring the role of Msx1 in regulating Wnt signaling modulators during early tooth development
Tooth development is a distinct embryological process that can be used as a model to study the multi-stage epithelial-mesenchymal interaction. The MSX1 transcription factor is essential in early tooth morphogenesis through bud-to-cap transitioning. Deleting the Msx1 gene causes tooth arrest at the bud stage in mice. In mice deficient in Msx1, Bmp4 mRNA expression in the dental mesenchyme is abated at E13.5 and E14.5. Alternatively, deleting the Bmp and Wnt modulator Sosdc1 acts as an impetus to forming supernumerary teeth as a result of dysregulated Bmp and Wnt signaling activity in the rudimentary dental tissues. These data confirm that Msx1 is upstream of the Wnt modulators (e.g., Dkk2 and Sosdc1) and regulates them independently during the bud-to-cap stage transition. However, the mechanism of Wnt modulator regulation by Msx1 in tooth development has not been determined and requires further studies.
Publications
- Profiles of Wnt pathway gene expression during tooth morphogenesis. Front Physiol 2024 10;14:1316635
- Spatial multi-omics reveals the role of the Wnt modulator, Dkk2, in palatogenesis. J Dent Res 2024 23:220345241256600
- Accelerating oral wound healing using bilayer biomaterial delivery of FTY720 immunotherapy. Adv Healthc Mater 2024 10:e2401480
Collaborators
- Justin L. Cotney, PhD, University of Connecticut School of Medicine, Farmington, CT
- Steven L. Goudy, MD, MBA, FACS, Emory University School of Medicine, Atlanta, GA
- James Iben, PhD, Molecular Genomics Core, NICHD, Bethesda, MD
- Sergey Leikin, PhD, Section on Physical Biochemistry, NICHD, Bethesda, MD