
Transcriptional Control of Cell Specification and Differentiation

- Jeffrey A. Farrell,
PhD, Stadtman Investigator, Head, Unit on Cell Specification and Differentiation - Morgan Prochaska, PhD, Research Specialist III
- Victor Naturale, PhD, Postdoctoral Intramural Research Training Award Fellow
- Michael Nunneley, PhD, Postdoctoral Intramural Research Training Award Fellow
- Gilseung Park, PhD, Postdoctoral Visiting Fellow
- Abhinav Sur, PhD, Postdoctoral Visiting Fellow
- Yalan Wu, BS, Predoctoral Medical Scientist Partnership Program Fellow
- Jackson Crane, BS, Postbaccalaureate Intramural Research Training Award Fellow
- Ella Segal, BS, Postbaccalaureate Intramural Research Training Award Fellow
Animals consist of a collection of cells with different shapes, structures, and functions, and this diversity of cell types is rebuilt from scratch by every embryo. The genetic programs that direct this process are the central mystery of developmental and regenerative biology. We are interested in how decisions about which cell type to adopt are controlled, and how genetic programs direct the morphological and functional specialization of different cells.
The single-cell revolution in developmental biology has given us new access and new tools to address these questions. I previously developed high-temporal-resolution, single-cell RNA–sequencing approaches to identify transcriptional trajectories, i.e., the ‘highways’ or the most likely paths through gene expression that cells take during development. From such data, we were able to identify the sequence of genes expressed by individual cell types during early development, which provides insight into the genetic programs that regulate cells’ choice of cell type and then their downstream functional transformations at a wider breadth than was previously achievable. Work in our lab focuses on more deeply exploring such processes, using the approaches we developed. Our lab combines single-cell genomics with imaging, genetic, and classical embryological approaches to investigate the genetic control of cell specification and differentiation during vertebrate embryogenesis. We focus on zebrafish (Danio rerio) embryos as a model system in which to study these questions, because their cell types and genetic regulation are extensively shared with humans; among vertebrates, they are easy to culture, image, and manipulate, both embryologically and genetically.
Transcriptional diversity during zebrafish development
A central quest in developmental biology is to understand the genetic programs that confer specific identities, morphologies, and behaviors to the many different cell types in a functioning animal. Our goal is to identify the cascades of gene expression within distinct cell types that drive specification (acquisition of an identity) and differentiation (acquisition of a particular function or morphology) and to understand their regulation. In 2023, we published a single-cell RNA-Seq (scRNA-Seq) atlas of wild-type development in zebrafish, spanning the first five days of development, that describes which genes are turned on in each cell type over time and created a heavily-used public resource with these data (Daniocell), which is used by investigators around the world to browse our data for their own specific research questions [Sur A, et al. Dev Cell 2023:58:3028].
This year, we made the Daniocell data even more useful and available to investigators worldwide by developing a software application (DaniocellDesktop) that enables reanalysis of these data without programming knowledge (Figure 1) [Reference 2]. Figures made with this software have appeared in publications and grant proposals and have helped drive forward the research projects of many.
Figure 1. DaniocellDesktop features
DaniocellDesktop allows users to plot (A) gene expression levels across cell types or developmental stages of interest, (B) plot gene co-expression, (C) define new cell-population definitions using their own criteria, and (D) perform differential gene-expression testing to find genes that are more highly or specifically expressed in particular cell populations.
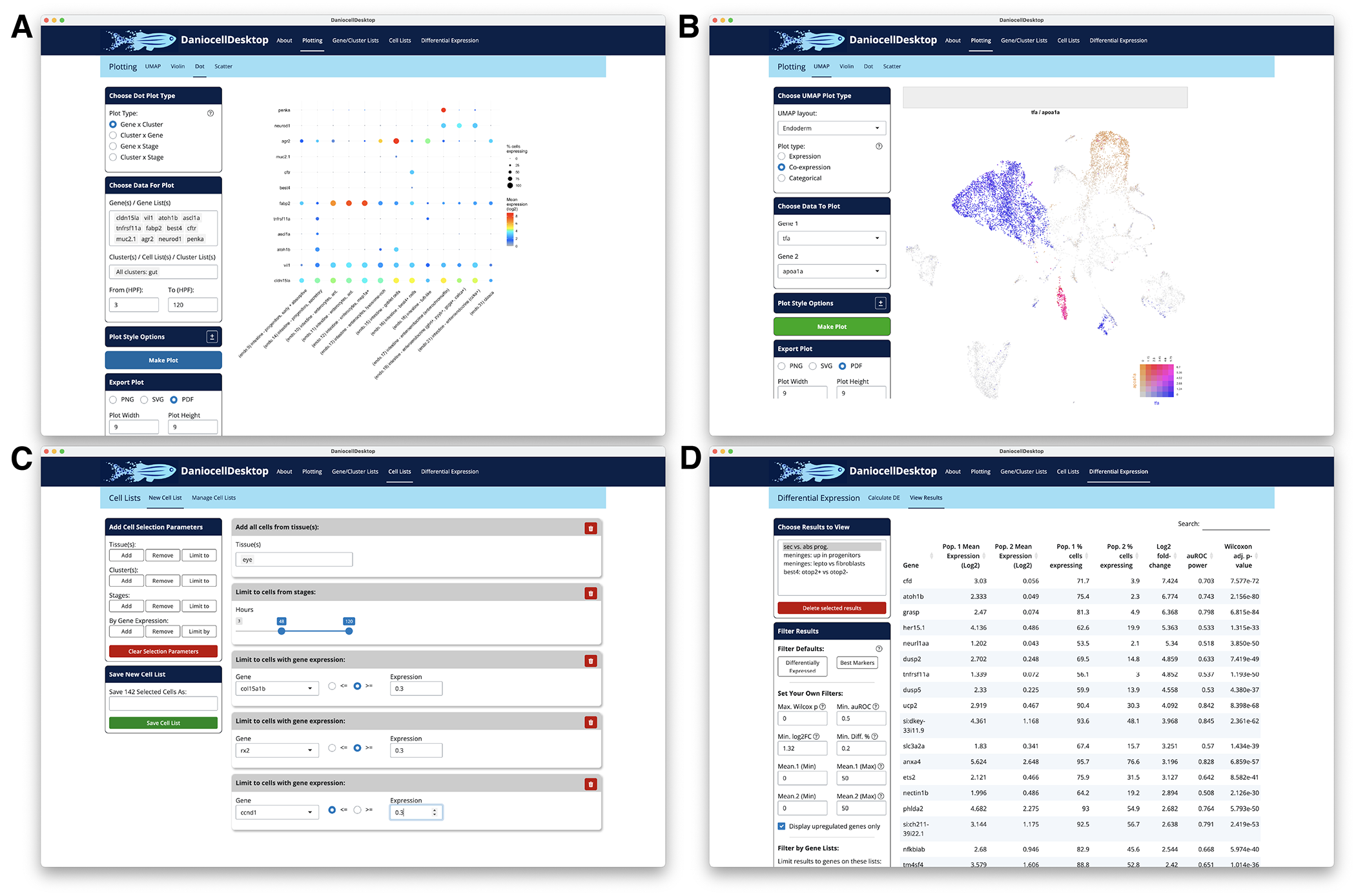
Figure 1. DaniocellDesktop features
DaniocellDesktop allows users to plot (A) gene expression levels across cell types or developmental stages of interest, (B) plot gene co-expression, (C) define new cell-population definitions using their own criteria, and (D) perform differential gene-expression testing to find genes that are more highly or specifically expressed in particular cell populations.
Additionally, while developmental trajectories identify the gene expression cascades that accompany the specification and differentiation of individual cell types, interpreting them biologically remains difficult. In a separate project, we helped develop MIMIR (Module Identification via Multi-source Integration for Reconstruction of differentiation) to identify gene modules that correspond to biological processes from scRNA-Seq trajectories by combining two info sources: similarity in gene expression dynamics and gene functional annotations [Reference 3]. Using MIMIR, we catalogued dozens of biological processes during the specification and differentiation of two embryonic structures with a shared progenitor: the notochord and prechordal plate, finding known and new biological processes in these tissues and discovering new gene associations (Figure 2). The approach revealed the anticipatory stimulation of the unfolded protein response (UPR) prior to cargo secretion. We found that both cell types express a shared 'core' UPR but also ‘cargo-specific’ UPRs that differ between tissues. To investigate whether this shared expression program has shared regulatory underpinnings in both tissues, we profiled embryos with loss-of-function and gain-of-function for UPR transcription factors (TFs). We found that even though there is a shared, 'core' UPR, those genes are actually activated by different UPR TFs in the two tissues. Additionally, in each tissue, the same TFs that activate the 'core' UPR also activate the 'cargo-specific' UPR genes. Moreover, activation of some ‘cargo-specific’ UPR genes requires a developmentally programmed co-factor. The study provides an approach to uncover biology in scRNA-Seq data from other cell types and tissues, offers a comprehensive view of cellular processes in notochord and prechordal plate development, and sheds light on cell type–specific regulation of shared processes.
Figure 2. Discovery of differential regulation of secretory behavior using MIMIR
MIMIR combines gene-expression information and gene functional annotation databases to identify biologically relevant gene-expression programs during development from single-cell transcriptional trajectories. We applied this to two embryonic cell populations (the notochord and hatching gland) that originate from the same progenitors to describe the biography of these cell types through the gene-expression programs. Both cell types are highly secretory, and our analysis identified shared and cell-type or cargo-specific secretory programs. We dissected the regulation of these programs using both loss-of-function and gain-of-function approaches to determine their regulation by the factors creb3l1, creb3l2, xbp1, and bhlha15 across these two cell types.
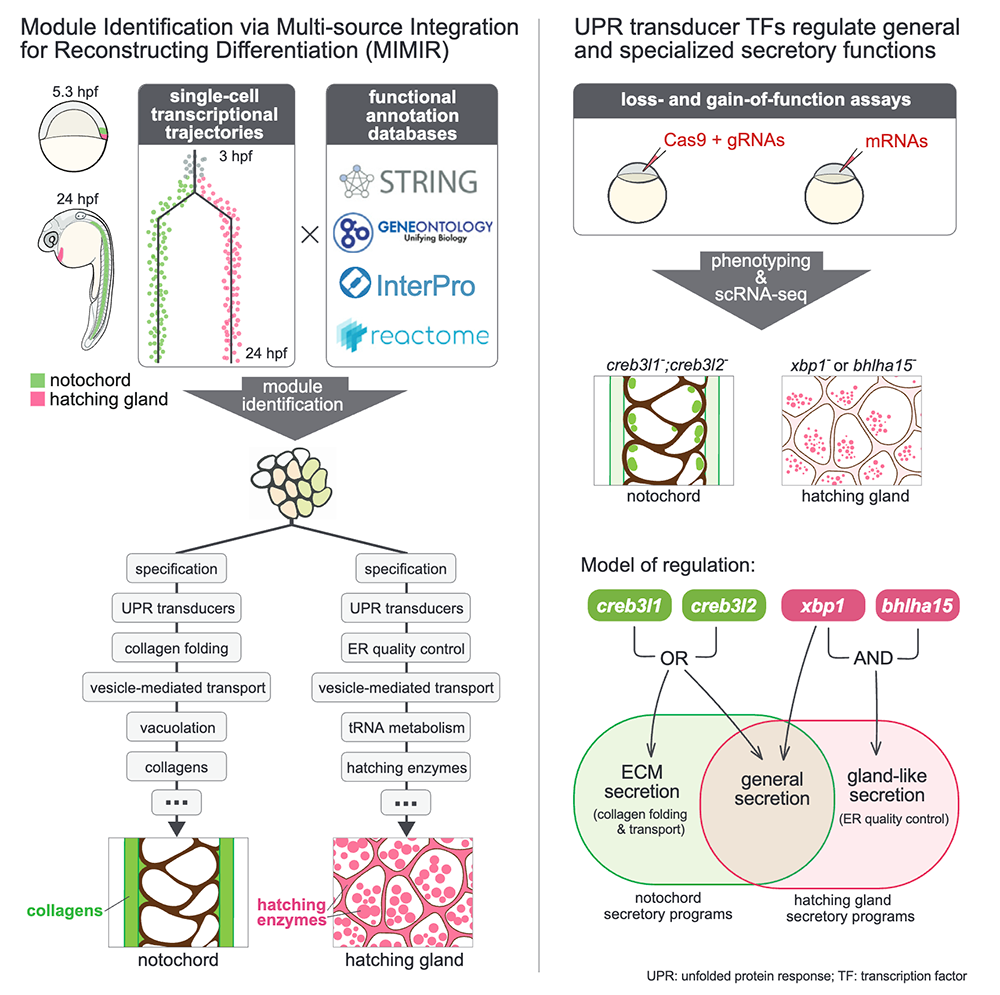
Figure 2. Discovery of differential regulation of secretory behavior using MIMIR
MIMIR combines gene-expression information and gene functional annotation databases to identify biologically relevant gene-expression programs during development from single-cell transcriptional trajectories. We applied this to two embryonic cell populations (the notochord and hatching gland) that originate from the same progenitors to describe the biography of these cell types through the gene-expression programs. Both cell types are highly secretory, and our analysis identified shared and cell-type or cargo-specific secretory programs. We dissected the regulation of these programs using both loss-of-function and gain-of-function approaches to determine their regulation by the factors creb3l1, creb3l2, xbp1, and bhlha15 across these two cell types.
We also collaborated with other zebrafish labs this year to use our single-cell RNA-Seq data and analysis expertise to help test or develop hypotheses for how several cell types develop. Our data helped test the gene-regulatory network that distinguishes auditory hair cells in otic structures that enable hearing from hair cells in the lateral line that detect mechanical disturbance or nearby motion [Reference 4]. We developed hypotheses for the progenitors and developmental regulation that give rise to leptomeningeal barrier cells in early development, cells that line blood vessels in the brain to help keep blood and cerebrospinal fluid isolated from each other [Reference 5]. Our group also contributed to analyses that identified zebrafish aerocytes in the gill and characterized their transcriptional similarity to mammalian lung aerocytes, a recently identified cell type that is specialized for gas exchange [Park JS, et al. bioRxiv 2025;doi:10.64898/2025.11.30.690480;preprint]. All these studies focused on cell types that are conserved between zebrafish and humans, where the ability to conduct rigorous in vivo experiments to determine how to create or manipulate these cells in zebrafish can then be translated to humans and thus contribute to medical therapies.
Development and function of best4+ cells
best4+ cells are a mysterious intestinal epithelial population, recently identified in humans (2019–2022), in zebrafish (2021–2023, by our group and others), and now in over a dozen vertebrates. The cells are dysregulated in disease: they are depleted in inflammatory bowel disease and their characteristic genes are upregulated in some colorectal cancer patients, which is predictive of better survival after surgery, suggesting that stimulation of their replenishment in the intestine may have therapeutic potential. However, it is unclear whether these changes are causes or consequences of disease. Additionally, given that the cells were recently discovered, it is unclear what function best4+ cells perform or how they develop. Since best4+ cells are evolutionarily lost in mice, we established zebrafish as a tractable in vivo model to observe, manipulate, and remove best4+ cells in an organismal context, where their interaction with other key tissues can be observed. We then used zebrafish to test how these cells develop and how they might function [Reference 1].
We dissected best4+ cell developmental regulation in vivo from birth to differentiation and specialization, focusing on factors that are evolutionarily conserved in best4+ cells. With lineage tracing, we demonstrated that best4+ cells arise from intestinal secretory progenitors that also give rise to other intestinal support cell types, such as goblet and enteroendocrine cells. Among secretory progenitors, Notch receptor is stimulated by the ligand Dll4 to mediate a decision between best4+ and enterochromaffin cells, a subtype of enteroendocrine cells. Notch signaling triggers expression of meis1b, a transcription factor that then confers best4+ cell identity. We found that best4+ cells have different gene expression in different regions of the intestine, and that they have correlated differences in their intracellular pH, which suggests that they may perform different functions in different regions of the intestine. We found that their regional diversification is regulated by the transcription factor Pbx3a. We generated transgenic lines that allow us to image best4+ cells over time and genetically manipulate them, which showed us that these cells make dynamic projections that extend towards the lumen of the intestine, as well as to other cells in the intestinal epithelium or surrounding the intestine, suggesting both sensory and communicative roles for these cells. These putative roles were further supported by experiments removing the cells in vivo and removing their spatial pattering. We additionally eliminated roles for these cells that have been proposed by others: we find that they are not required for regulation of intestinal pH and they do not absorb nutrients. Altogether, our work delineated a comprehensive best4+ cell developmental program, and we developed a genetic toolkit to dissect their function in vivo, both of which will be instrumental in understanding how best4+ cells are altered in disease and to stimulate the birth of new best4+ cells in patients (Figure 3).
Figure 3. A working model for best4+ cell development
Based on our developmental study, our working model for how best4+ cells are specified in development is that, initially, intestinal progenitors that do not receive Notch signaling become secretory progenitors. A subset of those secretory progenitors, which express the transcription factor Atoh1, then have Notch receptors stimulated by the ligand Dll4, which causes them to become best4+ cells instead of enterochromaffin cells. Notch triggers expression of the transcription factor Meis1, which confers their identity. The transcription factor Pbx3 then helps activate region-specific transcriptional programs that spatially and potentially functionally diversify best4+ cells.
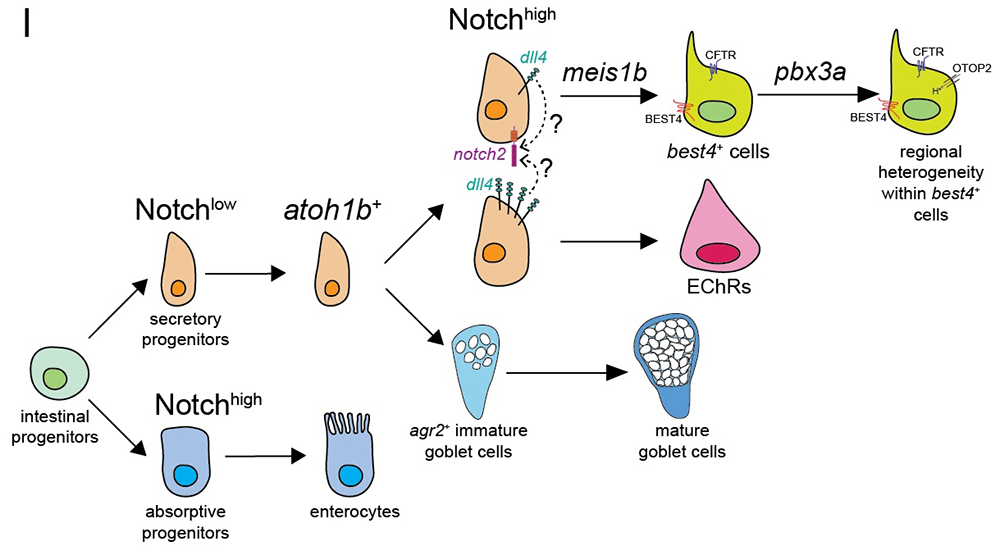
Figure 3. A working model for best4+ cell development
Based on our developmental study, our working model for how best4+ cells are specified in development is that, initially, intestinal progenitors that do not receive Notch signaling become secretory progenitors. A subset of those secretory progenitors, which express the transcription factor Atoh1, then have Notch receptors stimulated by the ligand Dll4, which causes them to become best4+ cells instead of enterochromaffin cells. Notch triggers expression of the transcription factor Meis1, which confers their identity. The transcription factor Pbx3 then helps activate region-specific transcriptional programs that spatially and potentially functionally diversify best4+ cells.
Additional Funding
- U.S.-Israel Binational Science Foundation 2023216 (to Jeffrey Farrell)
- K99/R00 Pathway to Independence Award (to Abhinav Sur)
- Jane Coffin Childs Memorial Fund Postdoctoral Fellowship (to Victor Naturale)
- NICHD Early Career Award (to Gilseung Park)
Publications
- Developmental regulation of intestinal best4+ cells. bioRxiv 2025 doi:10.64898/2025.12.17.694935;preprint
- DaniocellDesktop: for interactive reanalysis of wild-type zebrafish single-cell genomic data. Zebrafish 2025 22(6):230-232
- Gene module reconstruction identifies cellular differentiation processes and the regulatory logic of specialized secretion in zebrafish. Dev Cell 2025 60:1-18
- prdm1a drives a fate switch between hair cells of different mechanosensory organs. Nat Commun 2025 16(1):7662
- Anatomical and molecular characterization of the zebrafish meninges. bioRxiv 2025 doi:10.1101/2025.04.09.646894;preprint
Collaborators
- James Gagnon, PhD, University of Utah, Salt Lake City, UT
- Celina Juliano, PhD, University of California Davis, Davis, CA
- Tatjana Piotrowski, PhD, Stowers Institute, Kansas City, MO
- Michal Rabani, PhD, The Alexander Silberman Institute of Life Science, The Hebrew University of Jerusalem, Jerusalem, Israel
- Alexander F. Schier, PhD, Biozentrum, Basel, Switzerland
- Brant Weinstein, PhD, Section on Vertebrate Organogenesis, NICHD, Bethesda, MD
Contact
For more information, email jeffrey.farrell@nih.gov or visit https://www.nichd.nih.gov/research/atNICHD/Investigators/farrell.