
Transcriptional and Translational Regulatory Mechanisms in Nutrient Control of Gene Expression

- Alan G. Hinnebusch,
PhD, Head, Section on Nutrient Control of Gene Expression - Fan Zhang, MS, Senior Research Assistant
- Darren Fenton, PhD, Postdoctoral Fellow
- Ritu Gupta, PhD, Postdoctoral Fellow
- Rakesh Kumar, PhD, Postdoctoral Fellow
- Priyanka Mittal, PhD, Postdoctoral Fellow
- Poonam Poonia, PhD, Postdoctoral Fellow
- Sakshi Singh, PhD, Postdoctoral Fellow
- Anil Vijjamarri, PhD, Postdoctoral Fellow
- Brittany Benner, BS, Postbaccalaureate Fellow
- Aditi Chadha, BS, Postbaccalaureate Fellow
- Samuel Groysman, BS., Postbaccalaureate Fellow
- Hana Lee, BS, Postbaccalaureate Fellow
We study molecular mechanisms underlying the assembly, function, and regulation of translation initiation, using budding yeast as a model system to exploit its powerful combination of genetics and biochemistry. The translation initiation pathway produces an 80S ribosome bound to mRNA with methionyl initiator tRNA (tRNAi) base-paired to the AUG start codon in the ribosomal P site. The tRNAi is recruited to the 40S subunit in a ternary complex (TC) with GTP–bound eIF2 (eukaryotic translation initiation factor) to produce the 43S preinitiation complex (PIC) in a reaction stimulated by eIFs 1, 1A, 3, and 5. The 43S PIC attaches to the 5′ end of mRNA, facilitated by the cap-binding complex eIF4F (comprising eIF4E, eIF4G, and RNA helicase eIF4A) and poly(A)–binding protein bound to the poly(A) tail, and scans the 5′ UTR for an AUG start codon in preferred sequence context. Scanning is promoted by eIFs 1 and 1A, which induce an open conformation of the 40S, and by eIF4F and RNA helicase Ded1, which resolve RNA structures in the 5′ UTR. AUG recognition stabilizes a closed conformation of the PIC, dissociation of eIF1, highly stable P-site binding of tRNAi, and hydrolysis of the GTP bound to eIF2 stimulated by eIF5. Subsequent release of eIF2–GDP from tRNAi allows joining of the 60S subunit to form the 80S initiation complex.
eIF2 function is down-regulated by phosphorylation of its alpha subunit by protein kinase Gcn2 in response to amino acid starvation and other stresses that likely evoke stalling of ribosomes engaged in translation elongation, constituting one leg of the Integrated Stress Response, which is conserved throughout eukaryotes. Gcn2 exists as a latent enzyme that is recruited to and activated by stalled, frequently collided ribosomes, by the Gcn1/Gcn20 complex, and by the P-stalk complex of the 60S ribosome.
The initiation of protein synthesis is also regulated indirectly by the controlled degradation of mRNAs, which is generally initiated by removal of the poly(A) tail and 5′ m7G cap structure (decapping) and that differentially targets mRNAs depending on the functions of their encoded proteins and the metabolic needs of the cell. Decapping is enhanced by either slowly decoding or defective ribosomes or specific RNA–binding proteins, which recruit activators of the decapping enzyme Dcp1:Dcp2, including Dhh1, Pat1, Scd6, or Edc3, and is generally impeded by the poly(A)–binding protein (PABP) through its association with cap-bound eIF4F.
We also study fundamental mechanisms of transcriptional control of gene expression by analyzing the coordinate induction of over 100 amino-acid biosynthetic genes by activator Gcn4 during amino acid limitation. We and others showed previously that Gcn4 recruits multiple cofactor complexes, including SAGA, which contains the histone acetyltransferase (HAT) Gcn5, the chromatin remodeling (CR) complexes SWI/SNF and RSC, and the RNA Polymerase II (Pol II) Mediator, which in turn stimulate recruitment of general transcription factors (GTFs), including TATA–binding protein (TBP), and Pol II to promoter DNA to stimulate preinitiation complex (PIC) assembly. Seeking to identify all cofactors that mediate eviction of the –1 and +1 nucleosomes that occlude promoter DNA and impede binding of TBP and PIC assembly, we uncovered extensive cooperation among SWI/SNF, RSC, and a third CR, Ino80C, in evicting and repositioning promoter nucleosomes at genes activated by Gcn4 and other highly expressed genes in starved cells.
Yeast poly(A)–binding protein (Pab1) controls translation initiation in vivo primarily by blocking mRNA decapping and decay.
Poly(A)–binding protein (PABP, Pab1 in yeast) has been implicated in stimulating translation initiation and regulating mRNA decapping and decay by forming a closed-loop mRNP via interaction of PABP bound to the poly(A) tail with eIF4G bound to the m7G cap; however, PABP’s importance for promoting the translation or stability of individual transcripts is poorly defined in most eukaryotes, including budding yeast. Using an auxin-inducible pab1-AID degron mutant, we found that Pab1 depletion impairs yeast cell growth and polysome assembly and reduces bulk mRNA levels by about 40%. Strikingly, these phenotypes were suppressed by deleting DCP2 (dcp2∆), which encodes the catalytic subunit of the mRNA–decapping enzyme, implying that diminished mRNA abundance and global translation at limiting Pab1 results primarily from enhanced mRNA decapping and decay. Single-molecule sequencing of poly(A) tails (SM-PAT-Seq) revealed a shift to longer poly(A) tails on Pab1 depletion in DCP2, but not in dcp2∆ cells, which therefore likely results from preferential degradation of mRNAs with shorter poly(A) tails rather than slower deadenylation. Unlike findings in mammalian cells, where PABP depletion reduced mRNA abundance and protein synthesis but had little effect on translational efficiencies of individual mRNAs, we found that Pab1 depletion reprogrammed the translational efficiencies of about 1200 mRNAs and conferred corresponding changes in encoded protein levels. Importantly, these changes in translation were diminished or eliminated by deleting DCP2 and thus arise indirectly from reduced mRNA abundance on Pab1 depletion, possibly from altered ratios of mRNAs to eIF4F, preinitiation complexes, or other RNA–binding proteins. A subset of about 100 mRNAs still exhibit altered translational efficiencies on Pab1 depletion in dcp2∆ cells, suggesting a more direct stimulatory role for Pab1 in their translation. Nevertheless, our results indicate that assembly of the closed-loop mRNP via Pab1–eIF4G interaction is dispensable for wild-type translational efficiencies of the great majority of yeast mRNAs at normal transcript levels in vivo. Interestingly, histone mRNAs and proteins are preferentially diminished on Pab1 depletion, dependent on Dcp2, which is accompanied by activation of internal cryptic promoters in the manner expected for reduced nucleosome occupancies, revealing a new layer of post-transcriptional control over histone gene expression via Pab1.
Figure 1. Yeast poly(A)–binding protein (Pab1) controls translation initiation in vivo primarily by blocking mRNA decapping and decay.
Left panel: In wild-type (WT) yeast cells, poly(A)–binding protein (Pab1) forms a closed-loop mRNP by simultaneously interacting with the poly(A) tail and eIF4G of the eIF4E/eIF4G complex bound to the m7G cap (depicted in upper left schematic), which impedes decapping/degradation of most cellular mRNAs and indirectly stimulates translation of most mRNAs, promoting efficient polysome assembly (depicted in the inset) and enhancing the translational efficiencies (TEs) of a subset of transcripts. Histone mRNAs are preferentially stabilized by Pab1, ensuring histone protein expression and nucleosome densities within genic coding regions at levels sufficient to block RNA Pol II transcription from internal cryptic promoters. Right panel: Pab1 degradation induced by auxin treatment of a pab1-AID mutant disrupts closed-loop assembly and accelerates decapping/decay of most mRNAs, thereby diminishing bulk translation and reprogramming of TEs of over 1,000 mRNAs, all in a manner suppressed by eliminating the mRNA decapping enzyme (Dcp2). Pab1 depletion in cells with WT Dcp2 levels reduces histone mRNA/protein abundance, decreases nucleosome densities, and derepresses cryptic transcription from internal genic promoters.
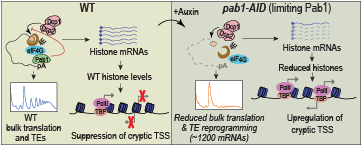
Figure 1. Yeast poly(A)–binding protein (Pab1) controls translation initiation in vivo primarily by blocking mRNA decapping and decay.
Left panel: In wild-type (WT) yeast cells, poly(A)–binding protein (Pab1) forms a closed-loop mRNP by simultaneously interacting with the poly(A) tail and eIF4G of the eIF4E/eIF4G complex bound to the m7G cap (depicted in upper left schematic), which impedes decapping/degradation of most cellular mRNAs and indirectly stimulates translation of most mRNAs, promoting efficient polysome assembly (depicted in the inset) and enhancing the translational efficiencies (TEs) of a subset of transcripts. Histone mRNAs are preferentially stabilized by Pab1, ensuring histone protein expression and nucleosome densities within genic coding regions at levels sufficient to block RNA Pol II transcription from internal cryptic promoters. Right panel: Pab1 degradation induced by auxin treatment of a pab1-AID mutant disrupts closed-loop assembly and accelerates decapping/decay of most mRNAs, thereby diminishing bulk translation and reprogramming of TEs of over 1,000 mRNAs, all in a manner suppressed by eliminating the mRNA decapping enzyme (Dcp2). Pab1 depletion in cells with WT Dcp2 levels reduces histone mRNA/protein abundance, decreases nucleosome densities, and derepresses cryptic transcription from internal genic promoters.
The mRNA decapping activators Edc3 and Scd6 act redundantly with helicase Dhh1 in nutrient-replete cells to post-transcriptionally repress starvation-induced pathways.
Degradation of most yeast mRNAs involves decapping by the Dcp1/Dcp2 complex. The Edc3 and Scd6 proteins have been implicated as decapping activators in budding yeast, but few native mRNAs were shown to depend on either protein for stimulating decapping and degradation. Scd6 has also been implicated as a translational repressor, but again, few native mRNAs were shown previously to be translationally derepressed in mutant cells lacking only Scd6. By combining RNA-Seq with ribosome profiling and global proteomics via TMT-mass spectrometry, we demonstrated that Scd6 and Edc3 function redundantly to repress the abundance or translation of a sizeable group of mRNAs, whose abundance is derepressed only in the mutant cells lacking both proteins. By interrogating our previous analyses of mutants lacking the decapping activators Dhh1 and Pat1, we discovered that mRNAs targeted for degradation or translational repression by Scd6/Edc3 are generally also targeted by Dhh1 or Pat1. An especially strong correspondence between mRNAs dysregulated in scd6∆edc3∆ and dhh1∆ mutants supports a recent proposal that Edc3 and Scd6 function interchangeably in recruiting Dhh1 to Dcp2 for activation of decapping, which we corroborated by co-immunoprecipitation analysis of native proteins in cell extracts. We further established that mRNAs preferentially repressed by Scd6/Edc3 encode proteins normally expressed at low levels on rich media and derepressed during growth on alternative carbon sources. These include the mitochondrial enzymes of oxidative phosphorylation, and we demonstrated elevated mitochondrial membrane potential and increased carbon flow from glucose into intermediates of the tricarboxylic acid cycle in mutants lacking Scd6/Edc3, Dhh1, or Pat1. Overall, we found that Scd6 and Edc3 are functionally redundant and cooperate extensively with Dhh1 and Pat1 in post-transcriptional repression of gene expression that promotes fermentation (glycolysis) over respiration in glucose-grown cells.
Transcriptional coactivator SAGA subunits Spt3 and Spt8 act directly and non-redundantly with TFIID in recruitment of the TATA–binding protein (TBP) in the Gcn4 transcriptome.
In budding yeast, TFIID and SAGA are the principal coactivator complexes that recruit TATA–binding protein (TBP) for assembly of preinitiation complexes at gene promoters, and the resident histone acetylation (HAT) activity of SAGA lodged in subunit Gcn5 enhances transcription via histone acetylation. Published evidence suggests that the majority of yeast genes (around 90%) require TFIID for efficient transcription, with only about 10% utilizing SAGA in addition to or instead of TFIID. It was concluded recently that the cohort of genes activated by transcriptional activator Gcn4 in amino acid–starved cells utilize only the HAT activity and not the TBP–recruitment function of SAGA and rely exclusively on TFIID for TBP recruitment. We found that eliminating the two TBP–interacting subunits of SAGA, Spt3 and Spt8, but not Gcn5, reduced TBP binding at most Gcn4 target genes in amino acid–starved cells. In contrast, eliminating Gcn5 but not Spt3 or Spt8, impaired histone acetylation and nucleosome eviction at highly expressed Gcn4 target genes. We further showed that depleting the key Taf1 subunit of TFIID strongly impairs TBP recruitment at most Gcn4 target genes only in cells devoid of Spt3 or Spt8. These results indicate a critical, non-redundant role for SAGA in TBP recruitment, apart from its HAT function, even in cells harboring wild-type TFIID. We obtained similar findings for the most highly expressed subset of genes previously shown to utilize SAGA or TFIID redundantly, dubbed “coactivator-redundant (CR)” genes. In contrast, highly expressed TFIID–dependent genes showed reduced TBP binding on depleting Taf1, even in cells with wild-type SAGA, and eliminating Spt3/Spt8 had little effect on TBP recruitment. These findings deepen our understanding of the relative importance of SAGA vs. TFIID in TBP recruitment, PIC assembly, and transcription of different classes of genes under stress conditions, highlighting the crucial role of SAGA in TBP recruitment via Spt3/Spt8 and the independent contribution of Gcn5 HAT activity for the Gcn4 transcriptome and other highly expressed CR genes in amino acid–starved cells.
Figure 2. Subunits Spt3 and Spt8 of transcriptional coactivator SAGA act independently of histone acetyltransferase Gcn5 and non-redundantly with coactivator TFIID in recruitment of TATA–binding protein at genes activated by transcription factor Gcn4 in amino acid–deprived cells.
Synthesis of the transcription factor Gcn4 is induced by amino acid starvation, increasing its binding to enhancer-like upstream activation sequences (UASs) in the promoters of starvation-induced amino-acid biosynthetic genes. Gcn4 recruits coactivator SAGA, and SAGA subunits Spt3 and Spt8 bind to TATA–binding protein (TBP) and deliver it to TATA sequences in the promoter, with TBP binding stabilized by the general transcription factor TFIIA. Although TBP is delivered to TATA/TFIIA independently by coactivator TFIID, the contribution of the SAGA subunits Spt3/Spt8 to TBP recruitment is required for robust transcriptional induction, even when TFIID is fully functional. Gcn5–mediated histone acetylation (red triangle) stimulates displacement of nucleosomes occluding the transcription start site (black arrow) to enhance transcriptional activation at many genes without affecting TBP occupancy.
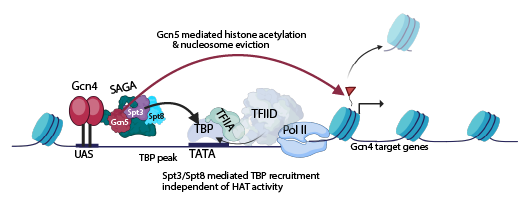
Figure 2. Subunits Spt3 and Spt8 of transcriptional coactivator SAGA act independently of histone acetyltransferase Gcn5 and non-redundantly with coactivator TFIID in recruitment of TATA–binding protein at genes activated by transcription factor Gcn4 in amino acid–deprived cells.
Synthesis of the transcription factor Gcn4 is induced by amino acid starvation, increasing its binding to enhancer-like upstream activation sequences (UASs) in the promoters of starvation-induced amino-acid biosynthetic genes. Gcn4 recruits coactivator SAGA, and SAGA subunits Spt3 and Spt8 bind to TATA–binding protein (TBP) and deliver it to TATA sequences in the promoter, with TBP binding stabilized by the general transcription factor TFIIA. Although TBP is delivered to TATA/TFIIA independently by coactivator TFIID, the contribution of the SAGA subunits Spt3/Spt8 to TBP recruitment is required for robust transcriptional induction, even when TFIID is fully functional. Gcn5–mediated histone acetylation (red triangle) stimulates displacement of nucleosomes occluding the transcription start site (black arrow) to enhance transcriptional activation at many genes without affecting TBP occupancy.
Enhancement of TBP recruitment to gene promoters by essential ATPase Mot1 varies with stress and gene-expression levels independently of promoter dependence on coactivators SAGA versus TFIID.
Previous studies had suggested that the ATPase Mot1 generally dissociates TBP from promoters that utilize SAGA to augment TBP recruitment in favor of TBP binding at genes reliant on TFIID for PIC assembly. Our findings show instead that promoters are dependent on Mot1 for high-level TBP occupancies when they are highly expressed, regardless of their dependence on SAGA or TFIID for TBP recruitment. Accordingly, TBP binding at condition-dependent SAGA–activated genes is enhanced by Mot1 when they are stress-induced, but antagonized by Mot1 when expressed at basal levels. Among bulk promoters expressed constitutively, Mot1 serves to redistribute TBP from weaker to stronger promoters regardless of their coactivator dependence. Our analysis also uncovered an unexpected response to Mot1 depletion whereby reductions in TBP occupancies at highly expressed/induced SAGA–dependent genes does not reduce their transcription levels. This uncoupling could arise from SAGA–mediated assembly of a reservoir of incomplete PICs containing TBP whose formation is not crucial for promoter activation per se, but may increase the rate or endpoint of induction. These findings substantially alter our understanding of the genome-wide role of Mot1 in regulating PIC assembly and its importance for mounting an effective transcriptional response to stress.
Publications
- Gcn2 structurally mimics and functionally repurposes the HisRS enzyme for the integrated stress response. Proc Natl Acad Sci USA 2024 121:e2409628121
- Yeast poly(A)-binding protein (Pab1) controls translation initiation in vivo primarily by blocking mRNA decapping and decay. Nucleic Acids Res 2025 53:gkaf143
- SAGA subunits Spt3 and Spt8 act directly and non-redundantly with TFIID in TBP recruitment in the Gcn4 transcriptome. Nucleic Acids Res. 2025 53:gkaf598
- Mot1 regulation of promoter binding by TBP varies with stress and gene expression levels independently of coactivator dependence. Proc Natl Acad Sci USA 2025 122:e2516926122
- Decapping activators Edc3 and Scd6 act redundantly with Dhh1 in post-transcriptional repression of starvation-induced pathways. eLife 2025 13:RP102287
Collaborators
- Miriam L. Greenberg, PhD, Department of Biological Sciences, Wayne State University, Detroit, MI
- Sunil Laxman, PhD, Institute for Stem Cell Science and Regenerative Medicine (DBT-inStem), Bengaluru, India.
- Zhenguo Lin, PhD, Department of Biology, Saint Louis University, St. Louis, MO
- Jon Lorsch, PhD, Laboratory on the Mechanism and Regulation of Protein Synthesis, NICHD, Bethesda, MD
- Neelam Sen, PhD, Jawaharlal Nehru University, New Delhi, India
- Anil Thakur, PhD, Regional Centre for Biotechnology, Faridabad, India
- Leos Valášek, PhD, Institute of Microbiology of the Czech Academy of Sciences, Prague, Czech Republic
- Jinwei Zhang, PhD, Laboratory of Molecular Biology, NIDDK, Bethesda, MD
Contact
For more information, email hinnebua@mail.nih.gov or visit https://www.nichd.nih.gov/research/atNICHD/Investigators/hinnebusch.