
Molecular Nature and Functional Role of Dendritic Voltage-Gated Ion Channels

- Dax Hoffman,
PhD, Head, Section on Molecular Neurophysiology and Biophysics - Jiahua Hu, PhD, Staff Scientist
- Lin Lin, PhD, Microbiologist
- Ying Liu, MD, Biologist
- Cole Malloy, PhD, Postdoctoral Fellow
- Meghyn Welch, PhD, Postdoctoral Fellow
- Ashley Pratt, BS, PhD Student
- Kailey Jerome, BS, Postbaccalaureate Fellow
The central nervous system (CNS) underlies all our experiences, actions, emotions, knowledge, and memories. With billions of neurons each firing hundreds of times per second, the complexity of the brain is stunning. To pare down the task of understanding something so complex, our research approach calls for studying the workings of a single central neuron: the pyramidal neuron from the CA1 region of the hippocampus. In humans, the hippocampus is essential for long-term memory and is among the first brain regions affected by epilepsy and Alzheimer’s disease. To understand how the hippocampus stores and processes information, we focus on the CA1 pyramidal neuron, one of its principal cell types. Each of these cells receives tens of thousands of inputs onto its dendrites, and it is commonly thought that information is stored by altering the strength of individual synapses (synaptic plasticity). Recent evidence suggests that the regulation of synaptic-surface expression of glutamate receptors can, in part, determine synaptic strength. However, the dendrites contain an abundance of ion channels that are involved in receiving, transforming, and relaying information in the dendrites, adding an additional layer of complexity to neuronal information processing.
We found that the A-type potassium channel subunit Kv4.2 is highly expressed in the dendritic regions of CA1 neurons in the hippocampus and, as one of the primary regulators of dendritic excitability, plays a pivotal role in information processing. Kv4.2 is targeted for modulation during the types of plasticity thought to underlie learning and memory. Moreover, we found that the functional expression level of Kv4.2 regulates the subtype expression of NMDA–type glutamate receptors, the predominant molecular devices controlling synaptic plasticity and memory. We are currently following up on these findings with more detailed investigations into the mechanisms of activity-dependent Kv4.2 regulation. In addition, we have begun to investigate the role of dendritic voltage-gated potassium and calcium channels in neuronal development and developmental disorders.
Role of voltage-gated ion channels in synaptic development and disease
Kv4.2 is an activity-dependent Ube3A substrate and contributes to synaptic plasticity and cognitive flexibility in Angelman syndrome.
Angelman syndrome (AS) is a severe neurodevelopmental disorder affecting 1 in 20,000 people, and is caused by the loss of function of imprinted genes on chromosome 15q11–13 or mutations in Ube3A. Ube3A (ubiquitin protein ligase E3A) is expressed exclusively from the maternal allele in hippocampal neurons and cerebellar Purkinje cells. Loss of Ube3A function leads to accumulation of target proteins, disrupting neuronal function. A TAP-MS (tandem affinity purification–mass spectrometry) screen identified Ube3A as a Kv4.2 binding protein. Follow-up studies by Jiahua Hu confirmed activity-dependent Kv4.2–Ube3A interaction, showing that Ube3A ubiquitinates Kv4.2 at residue K103, which is required for activity-induced Kv4.2 degradation.
In an AS mouse model, Kv4.2 protein levels and K+ currents are elevated in the hippocampus. Seizure-induced Kv4.2 degradation, which normally requires Ube3A, is absent in AS mice. Ube3A–mediated Kv4.2 ubiquitination is significantly reduced in AS hippocampi, further supporting the role of Ube3A in Kv4.2 degradation. Additionally, studies showed that seizure-induced Kv4.2 degradation occurs on dipeptidyl peptidase–like protein 6 (DPP6)–containing Kv4.2 complexes, requiring Kv4.2 phosphorylation at the T602/7 Pin1 site.
Patch clamp studies by Cole Malloy revealed deficits in mEPSC (miniature excitatory postsynaptic current) frequency and spike-timing–dependent long-term potentiation (LTP) in AS mice, which were rescued by crossing AS mice with conditional Kv4.2 knock-out (Kv4.2cKO) mice. Behavioral tests showed that some locomotion, nesting, and learning impairments in AS mice were also partially rescued in AS/Kv4.2cKO mice. In learning and memory tests, AS mice showed impairments in initial learning and reversal learning in an operant reversal test. However, the deficits in AS mice in reversal learning can be rescued by double knock-out mice. These findings reveal a novel Ube3A downstream pathway regulating plasticity and cognitive behaviors, and provide potential targets for the treatment of AS (see Image).
Figure 1. Working model of neuronal activity or seizure-induced Kv4.2–DPP6 complex remodeling, which may contribute to abnormal phenotypes in Angelman-syndrome (AS) mice.
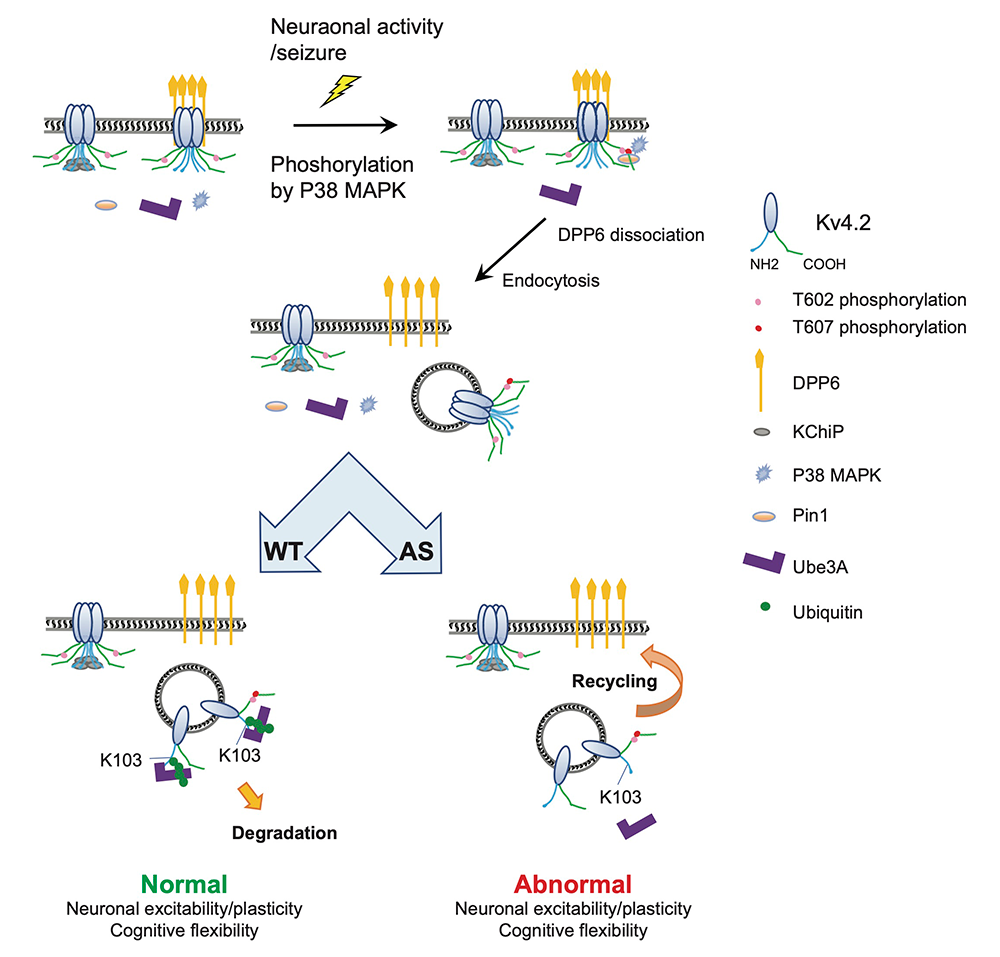
Figure 1. Working model of neuronal activity or seizure-induced Kv4.2–DPP6 complex remodeling, which may contribute to abnormal phenotypes in Angelman-syndrome (AS) mice.
Kv4.2 complex regulation and its role in cognitive flexibility
To address the role of the p38-Pin1–Kv4.2 pathway in neuronal excitability and circuit function, we developed a mutant knock-in mouse model with a Thr607 to Ala substitution at the activity-induced p38 phosphorylation site (T607 to A607; Kv4.2TA). We found that Kv4.2TA mice exhibit normal initial learning and memory in the Morris water maze and lever press, two tests of hippocampal-dependent learning and memory. However, they exhibited better 'reversal' learning in both tests than did wild-type (WT) mice. This improvement in reversal learning is indicative of an enhancement in cognitive flexibility. Cole Malloy is investigating the mechanisms behind enhanced cognitive flexibility in Kv4.2TA mice, the first mouse model with this phenotype. Focusing on synaptic differences between Kv4.2TA and WT mice, patch clamp electrophysiology in hippocampal CA1 pyramidal cells showed that Kv4.2TA mice have similar basal synaptic transmission and preserved long-term plasticity. However, they exhibit significant enhancement in the reversal of spike-timing–dependent long-term potentiation with low-frequency stimulation (LFS), suggesting a synapse state–dependent difference in synaptic plasticity attributable to altered Kv4.2 complex regulation.
Pharmacological manipulations during LFS revealed distinct mechanisms driving this metaplasticity. The NMDA antagonist 5-AP, which fully blocks depotentiation in WT mice, only partially blocks it (about 40%) in Kv4.2TA mice, indicating an additional mechanism in Kv4.2TA mice. The hypothesis is that altered metabotropic glutamate receptor (mGluR) signaling, specifically mGluR5, underlies this enhancement. Co-application of the mGluR5 antagonist MTEP with 5-AP fully rescues the depotentiation in Kv4.2TA mice, bringing it to WT levels, while MTEP alone fully prevents depotentiation in Kv4.2TA mice and only partially affects WT mice, suggesting that depotentiation in Kv4.2TA mice is primarily driven by mGluR5, while in WT mice, NMDA receptors are more dominant.
In summary, our study uncovers a novel metaplasticity mechanism in Kv4.2TA mice linked to cognitive flexibility, with implications for therapeutic strategies targeting neurodevelopmental disorders with cognitive flexibility impairments. It reveals a shift in the molecular drivers of depotentiation between Kv4.2TA and WT mice, likely because of impaired Kv4.2 complex trafficking during synaptic activity.
Interaction between mGluR5 and Kv4.2
Our previous research identified the metabotropic glutamate receptor 5 (mGluR5) within the Kv4.2 protein complex through TAP-MS analysis. Both mGluR5 and Kv4.2 are peri-synaptically localized. We hypothesized that mGluR5 traffics with Kv4.2 in response to neuronal activity. It is well documented that co-expression with DPP6 and Kv channel–interacting protein 2 (KChIP2) enhances Kv4.2 surface expression, which subsequently increases mGluR5 surface expression, suggesting a regulatory role of Kv4.2 on mGluR5 surface expression. To investigate internalization dynamics, we utilized a bungarotoxin (BTX) binding site–tagged Kv4.2 construct (BBS–Kv4.2) to label surface Kv4.2 in live HEK 293T cells co-transfected with hemagglutinin (HA)–tagged mGluR5. Under control conditions (4℃), both proteins were localized on the cell surface. Upon incubation at 37℃ to induce trafficking, internalized mGluR5 colocalized with Kv4.2, indicating potential co-trafficking of these proteins.
Behaviorally, Kv4.2TA mice demonstrated normal initial acquisition in the Morris water maze but exhibited improved reversal learning capabilities. In contrast, mGluR5 knockout (mGluR5KO) mice displayed impairments in both initial acquisition and reversal learning. Contextual fear conditioning and extinction tests further revealed that mGluR5KO mice have impaired extinction of contextual fear memory, which nullifies the enhanced extinction observed in Kv4.2TA mice, underscoring the essential role of mGluR5 in cognitive flexibility. Together, results from our studies show a synaptic correlate that accompanies enhanced contextual inhibitory learning in the form of a unique metaplasticity in the hippocampus, which is driven by mGluR5 in association with the Kv4.2 multimolecular complex.
Preso1 regulation of Kv4.2
Preso1 (also known as FRMPD4) is a multidomain post-synaptic scaffold protein highly expressed in the hippocampus. Mutations in the PRESO1 gene are associated with intellectual disability and cognitive decline in Alzheimer's disease patients. Previous studies have shown that Preso1 acts as a scaffold for mGluRs, recruiting proline-directed kinases that modulate receptor interaction with the regulatory protein Homer. Our research identifies Preso1 as a regulator of Kv4.2 channels in the hippocampus.
Using a Preso1 knockout (KO) mouse model, we observed a significant reduction (about 30%) in A-type potassium current (IA) in CA1 pyramidal neurons, leading to increased somatic and dendritic excitability, as evidenced by a higher neuronal firing frequency, a more hyperpolarized action-potential threshold, greater action-potential amplitude, lower rheobase, and shorter latency to fire an action potential.
Synaptic plasticity at hippocampal CA3-CA1 synapses was compromised, with notable deficits in both spike-timing–dependent long-term potentiation (LTP) and N-methyl-D-aspartate receptor (NMDAR)–dependent long-term depression (LTD). These findings highlight the critical role that Preso1 plays in maintaining hippocampal excitability and synaptic plasticity.
Behavioral assays revealed that Preso1-KO mice had impaired contextual memory and reversal learning, and increased initial learning (potentially because of heightened motivation) in lever press tasks. Kainic acid–induced seizure assessments showed no differences in overall seizure scores.
DPP6 and sleep/epilepsy in aging mice
Our studies demonstrated that aging DPP6 knockout (DPP6–KO) mice, a model related to Alzheimer's disease, exhibit circadian dysfunction and sleep disturbances. Using in vivo wireless implants to measure electroencephalogram (EEG), electromyogram (EMG), behavioral activity, and body temperature, we found that 12-month-old DPP6–KO mice had significantly reduced non-rapid eye movement (NREM) sleep and increased rapid eye movement (REM) sleep during the light-on phase, with heightened wake time. Sleep quality analyses indicated increased counts and transition times between sleep stages, pointing to fragmented sleep patterns. Overexpression of DPP6 in aging mice improved NREM sleep and decreased wake time, stabilizing sleep architecture and reducing fragmentation.
In terms of seizure activity, 12-month-old DPP6–KO mice exhibited significant epileptiform spikes and higher seizure scores following pentylenetetrazol (PTZ) induction. Immunofluorescence studies revealed a reduction in parvalbumin-positive (PV+) interneurons and degradation of perineuronal nets (PNNs) in the hippocampus, suggesting that these factors contribute to increased seizure susceptibility. Overall, our findings indicate that DPP6 plays a crucial role in regulating sleep and seizure susceptibility in aging mice, with potential therapeutic implications for Alzheimer's disease and epilepsy management.
Publications
- Activity-dependent degradation of Kv4.2 contributes to synaptic plasticity and behavior in Angelman syndrome model mice. Cell Rep 2025 44:115583
- R-type voltage-gated Ca2+ channels mediate A-type K+ current regulation of synaptic input in hippocampal dendrites. Cell Rep 2022 38(3):110264
- Alzheimer's disease/dementia-associated brain pathology in aging DPP6-KO mice. Neurobiol Dis 2022 174:105887
- Neuronal roles of the multifunctional protein dipeptidyl peptidase-like 6 (DPP6). Int J Mol Sci 2022 23(16):9184
- Activity-dependent isomerization of Kv4.2 by Pin1 regulates cognitive flexibility. Nat Commun 2020 11(1):1567
Collaborators
- Heather Cameron, PhD, Section on Neuroplasticity, NIMH, Bethesda, MD
- Thomas B. Friedman, PhD, Section on Human Genetics, NIDCD, Bethesda, MD
Contact
For more information, email hoffmand@mail.nih.gov or visit https://www.nichd.nih.gov/research/atNICHD/Investigators/hoffman.