
The Integration of Retrotransposon DNA and the Consequences to the Cell

- Henry L. Levin,
PhD, Head, Section on Eukaryotic Transposable Elements - Hyo Won Ahn, PhD, Research Fellow
- Angela Atwood-Moore, BA, Senior Research Assistant
- Abhishek Anand, PhD, Visiting Fellow
- SePil Lee, PhD, Visiting Fellow
- Alapani Mitra, PhD, Visiting Fellow
- Sonali Parida, PhD, Visiting Fellow
- Taeen Jidaan, BA, Postbaccalaureate Fellow
- Rebecca John, BA, Postbaccalaureate Fellow
Transposable elements (TEs) are prevalent in eukaryotes, where they constitute a large fraction of genomes, change genome structure, and provide regulatory sequences central to coordinating the expression of gene networks. In humans, TEs represent 50% of the genome. The dominant families of TEs are Long INterspersed Element-1 (LINE-1 or L1), which constitute 17% of the genome, and Alu Short Interspersed Elements (SINEs), which are mobilized by L1 proteins and constitute 10% of the genome. Given that TEs make up half of the human genome, it is not surprising that their regulatory features are abundant sources of tissue-specific promoter activity and are critical building blocks of gene-regulatory networks. Although the vast majority of TEs have lost mobility, each genome retains approximately 100 active copies. As a result, genome studies of human populations reveal many thousands of polymorphic TEs. These insertions have the potential to alter gene expression and impact health and disease. Despite the significant role TEs play in the architecture and function of the human genome, much is unknown about reactions responsible for L1 and Alu integration.
The relationship between the distribution of LEDGF along genes and the position of HIV-1 DNA integration
HIV-1 integration occurs across actively transcribed genes, a specificity that is the result of the interaction of chromatin factor LEDGF with IN (integrase). Our understanding of HIV-1 integration is incomplete, in part because the cellular function of LEDGF is unclear. Although LEDGF was originally isolated as a co-activator that stimulates promoter activity in purified systems, this model is inconsistent with LEDGF–mediated integration across gene bodies and with data indicating that LEDGF promotes transcriptional elongation. To clarify the cellular roles of LEDGF and its function in integration, we generated a highly specific map of its chromatin association using ChIP-Seq. We contended with the long-standing problem that antibodies specific for LEDGF have not been successful in ChIP-Seq experiments. By CRIPSR editing of HEK293T cells, we scarlessly introduce a 3XFLAG tag to the 3′ end of PSIP1, the native LEDGF gene. Surprisingly for a protein that mediates integration across gene bodies, we observed pronounced peaks of LEDGF at the 5′ end of transcription units, which matched the peaks of RNA Pol II and H3K4me3 at the transcription start sites (TSSs) of active promoters (Figure 1A). Using ChIP-Seq, we also observed that levels of RNA Pol II at promoters were reduced in the absence of LEDGF, demonstrating that LEDGF does recruit RNA Pol II (Figure 1B).
Figure 1. LEDGF is associated with TSSs (transcription start sites) where it recruits RNA Pol II.
A. Example of tracks showing enrichments of RNA Pol II, H3K4me3, MLL1, and LEDGF/p75 in cells with depleted MLL1. B. Metagene plot showing reduction of RNA Pol II in LEDGF/p75 KO cells. C. Metagene plot showing reduction of LEDGF/p75 at TSS when the IBD is removed.
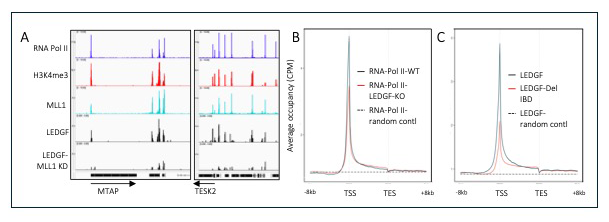
Figure 1. LEDGF is associated with TSSs (transcription start sites) where it recruits RNA Pol II.
A. Example of tracks showing enrichments of RNA Pol II, H3K4me3, MLL1, and LEDGF/p75 in cells with depleted MLL1. B. Metagene plot showing reduction of RNA Pol II in LEDGF/p75 KO cells. C. Metagene plot showing reduction of LEDGF/p75 at TSS when the IBD is removed.
Efforts to understand how LEDGF is recruited to promoters tested the function of factors that bind to LEDGF, such as the histone methyltransferase MLL1. ChIP-Seq showed that MLL1 peaks matched the positions of LEDGF at promoters and, importantly, when levels of MLL1 were reduced with shRNA (small interfering RNA hairpin), the peaks of LEDGF at promoters were significantly reduced (Figure 1A). Interestingly there is a reciprocal relationship, as cells lacking LEDGF exhibited a reduction of MLL1 at active promoters. These experiments provided insight into the function of LEDGF in transcription. Measures of LEDGF association at individual TSSs showed strong correlation with amounts of integration in the downstream transcribed sequences, indicating that MLL1, by recruiting LEDGF to TSSs, plays an important role in the genome-wide pattern of integration.
LEDGF possesses an N-terminal PWWP domain, which is known to interact with histone H3K36me3, an epigenetic mark found across transcribed sequences. The current model of integration is that the PWWP domain (a 100–150 amino acid structure found in eukaryotic proteins involved in DNA methylation, repair, and transcription) is responsible for directing LEDGF directly to the bodies of genes being actively transcribed. This tethers integrase to transcribed sequences, where it inserts HIV-1 cDNA. With ChIP-Seq experiments, we mapped the chromatin binding of LEDGF lacking the PWWP domain and found that no changes occurred in binding locations, results that were surprising because crude measures of chromatin association indicated that PWWP was important for binding. We tested the importance of H3K36me3–modified nucleosomes in directing integration with our collaborator Alan Engelman, who mapped integration sites in cells that lack H3K36me3. Importantly, we found that the frequencies and specificities of integration in genes were unchanged.
To determine which domains of LEDGF may direct its chromatin binding to TSSs, we expressed LEDGF lacking the integrase binding domain (IBD), the region that associates with integrase as well as with several cellular factors involved in transcription elongation. Importantly, we found that LEDGF lacking the IBD had far less enrichment at TSSs (Figure 1C). MLL1 is one of the factors that directly associates with the IBD and, together with the role of MLL1 in recruiting LEDGF to TSSs, these data indicate that MLL1 plays a key role in the chromatin association of LEDGF at TSSs. Based on these data, we propose a new model of LEDGF–mediated HIV-1 integration. LEDGF is recruited to active promoters by MLL1 and subsequently associates with the RNA Pol II elongation complex, which travels across transcription units to effect HIV-1 integration (Figure 2).
Figure 2. Model for the distribution of integration across actively transcribed genes
At TSSs MLL1 and LEDGF associate and recruit RNA Pol II. The IBD (integrase binding domain) of LEDGF is bound by MLL1 blocking the association of IN (integrase). During elongation, the IBD is available to interact with IN, allowing integration (inverted U) to occur across transcribed sequences.
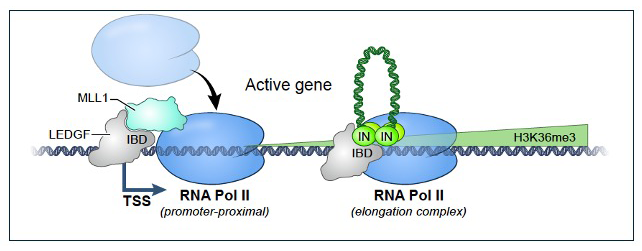
Figure 2. Model for the distribution of integration across actively transcribed genes
At TSSs MLL1 and LEDGF associate and recruit RNA Pol II. The IBD (integrase binding domain) of LEDGF is bound by MLL1 blocking the association of IN (integrase). During elongation, the IBD is available to interact with IN, allowing integration (inverted U) to occur across transcribed sequences.
L1 retrotransposition and the mechanism that causes 95% of insertions to have severe 5′ truncations.
The human genome contains about 500,000 copies of the non–LTR (long-terminal repeat) retrotransposon L1, most of which are inactive. L1 ORF2p (a protein essential for L1 retrotransposition) possesses endonuclease and reverse transcriptase activities that generate a single-stranded nick at the site of insertion and, at this position, perform target-primed reverse transcription. However, in 95% of L1 insertions, 5′ truncations occur that remove promoter and most of the protein coding sequences. 5′ truncation has played a significant role in shaping the 3-gigabase genome, which would, in the absence of 5′ truncation, have expanded dramatically to be 15 gigabases. We recently surveyed the frequencies of 5′ truncation of L1 elements in primate and murine evolution [Reference 1].
Little is known about the mechanism of truncation. To identify factors responsible for 5′ truncation, we developed an assay that measures both truncated and full-length insertions. We made a dual reporter system with GFP (green fluorescent protein) in the 3′ UTR and mCherry (red fluorescent protein) in the 5′ UTR of L1, where both genes are disrupted by an intron that is in the antisense orientation relative to L1 transcription (Figure 3). The reporters can only be expressed after splicing, reverse transcription, and integration of L1 into the genome. Full-length L1 insertions express red and green reporters, but if 5′-truncated, only the green reporter is active. Comparing the ratios of HEK293T cells expressing dual fluorescence with cells that only express green fluorescence, using FACS (fluorescence-activated cell sorting), reveals that full-length insertions represent approximately 5% of all de novo integration. We sequenced full-length and truncated insertions in HEK293T cells produced by the dual reporter to confirm the presence or absence of truncations in integrated L1.
Figure 3. Dual reporter of L1 transposition measures full-length integration.
The dual reporter of L1 (non-LTR retrotransposon) retrotransposition includes mCherry in the 5′ UTR and GFP in the 3′ UTR. Both fluorescent genes are disrupted with an intron that prevents their expression. The expression of mCherry (red fluorescent protein) and GFP (green fluorescent protein) occur only after the introns are spliced from the L1 RNA, the RNA is reverse transcribed, and the DNA is integrated. Insertions that are 5′ truncated are GFP+, while full-length insertions are both GFP+ and mCherry+.
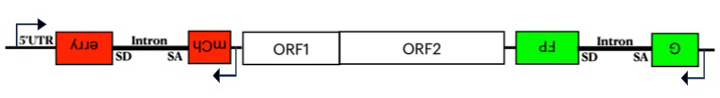
Figure 3. Dual reporter of L1 transposition measures full-length integration.
The dual reporter of L1 (non-LTR retrotransposon) retrotransposition includes mCherry in the 5′ UTR and GFP in the 3′ UTR. Both fluorescent genes are disrupted with an intron that prevents their expression. The expression of mCherry (red fluorescent protein) and GFP (green fluorescent protein) occur only after the introns are spliced from the L1 RNA, the RNA is reverse transcribed, and the DNA is integrated. Insertions that are 5′ truncated are GFP+, while full-length insertions are both GFP+ and mCherry+.
In efforts to screen large numbers of candidate genes for a role in 5′ truncation, we made another version of the reporter with an inducible promoter, and integrated the entire construct in HEK293T cells, using a Piggyback transposon delivery system. For genetic uniformity, we made single-cell–derived clonal populations from these cells. In one clonal cell line, we screened approximately 95 candidate genes for contributions to 5′ truncation using siRNA knockdown methods. CRISPR–mediated knockout of one candidate MOV10 confirmed a two-fold increase in full-length insertions. MOV10 is a 5′ to 3′ RNA helicase shown to associate with L1 RNA. We tested whether MOV10 associates with the L1 RNA using PAR-CLIP, a crosslinking method that identifies specific RNA sequences precipitated by a specific protein such as MOV10. Importantly, we found significant association of MOV10 along the 3′ region of L1 RNA, which is in the area where 5′ truncation occurs. We tested whether MOV10 restricts reverse transcription in in vitro assays of L1 particles purified from HEK293T cells. We found that L1 particles from cells that overexpress MOV10 produce reverse transcripts that are shorter than cells overexpressing MOV10 with a mutation that inactivates the helicase. These data suggest a collision model in which MOV10 bound to the 3′ end of L1 RNA obstructs the progress of ORF2 during reverse transcription (Figure 4).
Figure 4. The collision model for 5′ truncation of L1 integration
MOV10 bound to the 3′ end of L1 RNA obstructs the progress of ORF2 as it reverse transcribes L1 RNA. The result is the disruption of reverse transcription and truncation of the 5′ end of the insertions.
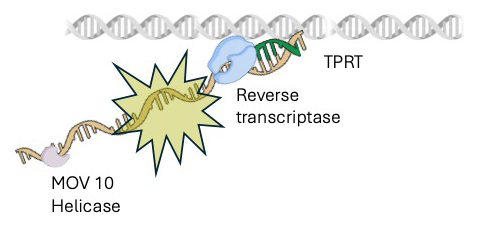
Figure 4. The collision model for 5′ truncation of L1 integration
MOV10 bound to the 3′ end of L1 RNA obstructs the progress of ORF2 as it reverse transcribes L1 RNA. The result is the disruption of reverse transcription and truncation of the 5′ end of the insertions.
Retrotransposon insertions associated with the risk of neurologic and psychiatric diseases
Mental disorders affected about 970 million people worldwide in 2017. In 2020, 21% of adults in the United States suffered from some form of mental illness. Such diseases thus cause great social and economic burden. Studies of identical twins show that the heritability of diseases such as attention-deficit hyperactivity disorder (ADHD), autism spectrum disorder (ASD), bipolar disorder (BIP), and schizophrenia is extremely high, ranging from 74% to 81%. Because of the complexity of the mammalian nervous system, the genetic and cellular etiology of such diseases remains largely unclear. Progress in genetic methodology has provided the potential to identify mechanisms that underlie the diseases. One approach that has successfully identified important disease loci is genome-wide association studies (GWAS). However, in the cases of neurologic and major psychiatric disorders, GWAS have identified large numbers of loci, each associated with small increases in risk. Importantly, there is extensive overlap of the loci that contribute to major psychiatric disorders, indicating that related molecular mechanisms may underlie distinct clinical phenotypes.
TASs (trait-associated single-nucleotide polymorphisms [SNPs]) of GWAS are genetic tags identifying a genomic region that contains the causal mutation(s) and that lead to increased disease risk. Limits on the design of GWAS typically prevent such studies from identifying causal gene alleles. Thus, determining causal variants remains the most challenging and rate-limiting but also the most important step in defining the genetic architecture of diseases. The vast majority of GWAS TASs lie in intergenic or intronic regions and therefore do not alter coding sequence. For such SNPs to be causal, they would likely have regulatory effects on transcription. Structural variants, such as rearrangements, copy number variants, and transposable element (TE) insertions, constitute a substantial and disproportionately large fraction of the genetic variants found to alter gene expression.
In humans, the dominant families of TEs are long interspersed element-1 (LINE-1 or L1) and Alu elements, which are short interspersed elements (SINEs) and are mobilized by L1. TEs readily alter gene expression because they have evolved various sequences that act on enhancers. Given that TEs make up approximately 50% of the human genome, it is not surprising that their regulatory features are abundant sources of tissue-specific promoter activity.
Relatively recent TE insertions can proliferate in the population and become common alleles. The 1000 Genomes Project described genetic variation of diverse human populations by sequencing whole genomes of 2,504 individuals. The extensive survey of genetic variation detected 17,000 polymorphic insertions of TEs, which have the potential to alter gene expression and affect common disease risk. There may be functional consequences of common TE insertion variants that affect common disease risk. Some common polymorphic TEs have been implicated at disease loci detected by GWAS. Common polymorphic Alu (short transposable elements) insertions occur disproportionately near disease loci of GWAS, underscoring the fact that Alu insertions are potential causative variants.
Given the difficulty in identifying genetic variants responsible for neurologic and psychiatric disorders and the regulatory capacity of TEs, we tested whether polymorphic TEs are potential causative variants of such diseases. We analyzed 593 GWAS of neurologic and psychiatric diseases, which in total reported 753 TASs. From the 17,000 polymorphic TEs, we found that 76 were in linkage disequilibrium (LD) with TASs, indicating that the TEs were among the variants with the potential to be causative. We extended our analysis by evaluating each candidate TE for a role in altering expression of proximal genes. In one approach, we investigated whether polymorphic TEs could disrupt regulatory sequences, as annotated with the epigenomic data of the NIH Roadmap Epigenomics Consortium. In all, we identified 10 polymorphic TEs to examine further as causal candidates because they were positioned in enhancer, promoter, heterochromatin, or transcribed sequences present in neurologic tissues.
We hypothesized that polymorphic TEs can have a causal relationship with the risk of psychiatric and neurologic disorders by altering expression of genes in cis. For evidence of altered gene expression, we queried the Genotype-Tissue Expression (GTEx) database, which contains expression data for 948 donors across 54 tissues. GTEx readily identifies changes in tissue-specific gene expression associated with loci-specific genetic variation. SNPs in LD with a query gene are identified as eQTLs (expression quantitative trait loci) if the genetic loci with the variants are significantly associated with altered expression of a gene in a specific tissue. We found that 31 of the TASs linked to TEs were variants that are associated with changes in expression of one or more adjacent genes within regions of the brain.
Having identified a number of polymorphic Alu elements that are significantly associated with disease risk detected by GWAS and that are correlated with altered gene expression in neurologic tissues by eQTL analysis, we developed a luciferase reporter assay to test whether the insert sequences in the context of flanking sequence can influence transcription activity. We measured the impact of candidate Alu and flanking sequences on the function of a minimal promoter in NCRM-1 (human neural stem cells). Of six candidate Alu insertions evaluated for their impact on promoter activity, we found that five significantly altered the expression of luciferase. Taken together, we identified 10 polymorphic TE insertions that are potential candidates on par with other variants for having a causal role in neurologic and psychiatric disorders.
Additional Funding
- FY2024 Office of AIDS Research
Publications
- The 5' truncation of retrotransposon L1: a process of genome integrity. Genetics 2025 231:iyaf202
- The relationship between the distribution of LEDGF along genes and positions of HIV-1 DNA integration. mBio 2026 in press
Collaborators
- Kathleen Burns, MD, PhD, Dana-Farber Cancer Institute, Boston, MA
- Ryan Dale, MS, PhD, Bioinformatics and Scientific Programming Core, NICHD, Bethesda, MD
- Alan Engelman, PhD, Dana-Farber Cancer Institute, Boston, MA
- Caroline Esnault, PhD, Bioinformatics and Scientific Programming Core, NICHD, Bethesda, MD
- Avindra Nath, MD, PhD, Division of Neuroimmunology & Neurovirology, NINDS, Bethesda, MD
- Mikel Zaratiegui, PhD, Rutgers, The State University of New Jersey, Piscataway, NJ
Contact
For more information, email henry_levin@nih.gov or visit https://www.nichd.nih.gov/research/atNICHD/Investigators/levin.