
The Role of Endogenous Retroviruses and KRAB–ZFP Genes in Mammal Development and Evolution

- Todd S. Macfarlan,
PhD, Head, Section on Mammalian Development and Evolution - Dawn Watkins-Chow, PhD, Staff Scientist
- Sherry Ralls, BA, Biologist
- Melania Bruno, PhD, Visiting Fellow
- Jinpu Jin, PhD, Visiting Fellow
- Peter Lindner, MD, Visiting Fellow
- Anna Dorothea Senft, PhD, Visiting Fellow
- Rachel Cosby, PhD, Postdoctoral Intramural Research Training Award Fellow
- Jada Gonzales, BS, Postbaccalaureate Fellow
- Sharaf Maisha, BS, Postbaccalaureate Fellow
A central goal of the NICHD is to understand human development and improve reproductive health. In the Section on Mammalian Development and Evolution, we support this mission by studying the genetic and molecular changes that allowed mammals to evolve and how disruptions in these processes lead to human developmental disorders.
Although much is known about how mothers and fetuses interact during pregnancy, we still know surprisingly little about the genetic innovations that gave rise to placental mammals more than 100 million years ago, or about the mechanisms that continue to generate differences among mammalian species. One compelling idea is that ancient viruses that inserted themselves into the genomes of our ancestors played a key role in evolution. Rather than being purely harmful, these viral sequences contributed new genes and regulatory elements that helped shape mammalian traits, including placental development and the expansion and increased complexity of the brain.
Our work focuses on how these viral remnants, called endogenous retroviruses (ERVs), influence gene regulation during development. This led us to study a large and rapidly evolving family of genes known as KRAB zinc-finger proteins (KZFPs), which are the most abundant gene regulators in mammalian genomes. We hypothesize that KZFPs expanded as a defensive response to repeated viral invasions, evolving to keep these sequences under control. Supporting this idea, most KZFPs bind to these elements, and their loss in mice leads to abnormal gene activation. Furthermore, we have demonstrated that several ancient KZFPs and the sequences they interact with were likely repurposed to control essential developmental programs, contributing to mammalian innovations.
We are now investigating how this long-standing genetic “arms race” shaped mammalian evolution and how mutations in KZFPs contribute to human disease. We are also exploring new roles for KZFPs beyond gene silencing, including how the ancestral KZFP PRDM9 (see below) controls the placement of genetic recombination during egg and sperm formation. Through a partnership with Shady Grove Fertility Centers, we aim to build on our recent findings linking PRDM9 variation to a substantial fraction of human infertility cases, with the goal of improving diagnosis and reproductive outcomes.
Kruppel-associated box zinc-finger proteins (KRAB–ZFPs)
Kruppel-associated box zinc-finger (ZF) proteins (KRAB–ZFPs) are rapidly evolving transcriptional repressors, which emerged in a common ancestor of coelacanths, birds, and tetrapods; they constitute the largest family of transcription factors in mammals (estimated to be several hundred in mice and humans). Each species has its own unique repertoire of KRAB–ZFPs, with some shared by closely related species and others specific to each species. Remarkably, there was an explosion of KRAB–ZFP genes in the earliest mammals, many of which have been retained by purifying selection, but the function of these (as well as of the hundreds of species-restricted KRAB–ZFPs) have been largely unexplored. KRAB–ZFPs consist of an N-terminal KRAB domain, which binds to the co-repressor KAP1, and a variable number of C-terminal C2H2 ZF domains that mediate sequence-specific DNA binding. KAP1 interacts directly with the KRAB domain, which recruits the histone methyltransferase (HMT) SETDB1 and heterochromatin protein 1 (HP1) to initiate heterochromatic silencing. Several lines of evidence point to a role for the KRAB–ZFP family in ERV silencing. First, the number of C2H2 ZF genes in mammals correlates with the number of ERVs. Second, the KRAB–ZFP protein ZFP809 was isolated based on its ability to bind to the primer binding site for proline tRNA (PBSpro) of murine leukemia virus (MuLV). Third, deletion of the KRAB–ZFP co-repressors Trim28 or Setdb1 leads to activation of many ERVs. We therefore began a systematic interrogation of KRAB–ZFP function as a potential adaptive repression system against ERVs.
For this purpose, we have been focusing on: (1) fully annotating the KRAB–ZFP gene repertoire in mice; and (2) systematically exploring KRAB–ZFP gene function. We used a combination of long-read sequencing methods (Nanopore ultra-long reads and PacBio Hifi) to generate near–T2T (telomere-to-telomere) genome assemblies. With the further aid of long-read mRNA-Seq, we performed comprehensive annotations of KZFP genes in both lab mice and wild strains. Our data show that waves of TE (transposable element)/ERV integration are a driving force of KZFP gene innovation and adaptation within KZFP gene clusters. We also began a systematic analysis of KRAB–ZFPs using a medium-throughput ChIP-Seq screen and functional genomics of KRAB–ZFP clusters and individual KRAB–ZFP genes. Our ChIP-Seq data demonstrate that the majority of recently evolved KRAB–ZFP genes interact with and repress distinct and partially overlapping ERVs and other retrotransposons targets. The hypothesis is strongly supported by the distinct ERV reactivation phenotypes we observed in mouse ESC (embryonic stem cell) lines lacking one of five of the largest KRAB–ZFP gene clusters. Furthermore, KRAB–ZFP cluster knockout (KO) mice are viable, but have elevated rates of somatic retrotransposition of specific retrotransposon families, providing the first direct genetic link between KRAB–ZFP gene diversification and retrotransposon mobility. In contrast to the young (species-restricted) KRAB–ZFPs, we found that the older KRAB–ZFPs (which are conserved across mammals) bind to genetic loci that have themselves undergone regulatory innovations during evolution. By systematically studying these KRAB–ZFP genes and their targets, we are uncovering regulatory innovations unique to placental mammals.
CTCF barrier–breaking by ZFP661 promotes protocadherin diversity in mammalian brains.
Mammalian brains are larger and more densely packed with neurons than those of reptiles, but the genetic mechanisms underlying the increased connection complexity amongst neurons are unclear. The expression diversity of clustered protocadherins (Pcdhs), which is controlled by CTCF (CCCTC [DNA sequence]–binding factor) and cohesin, is crucial for proper dendritic arborization and cortical connectivity in vertebrates. We identified a highly conserved and mammalian-restricted KRAB–ZFP, ZFP661, which binds antagonistically at CTCF barriers at the Pcdh locus, preventing CTCF from trapping cohesin. ZFP661 balances the usage of Pcdh isoforms and increases Pcdh expression diversity. We demonstrated that loss of the gene Zfp661 causes cortical dendritic arborization defects and autism-like social deficits in mice. The human ZFP661 ortholog, called ZNF2, likewise binds to conserved sequences at the PCDH locus in humans, and copy number variations of ZNF2 are associated with autism. Our study reveals both a novel mechanism that regulates the trapping of cohesin by CTCF and a mammalian adaptation that promoted Pcdh expression diversity to accompany the expanded mammalian brain.
Figure 1. ZFP661 binds adjacent to a small subset of CTCF sites within loop anchors.
A. ZFP661 binding peaks overlap with a small subset of CTCF binding peaks.
B. Heat maps of ChIP-Seq signal across ZFP661 binding sites indicate strong overlap with CTCF and the cohesin subunit Rad21.
C. ZFP661 binding is found adjacent to CTCF binding motifs, within loop anchors, at the same position at which cohesin is typically trapped.
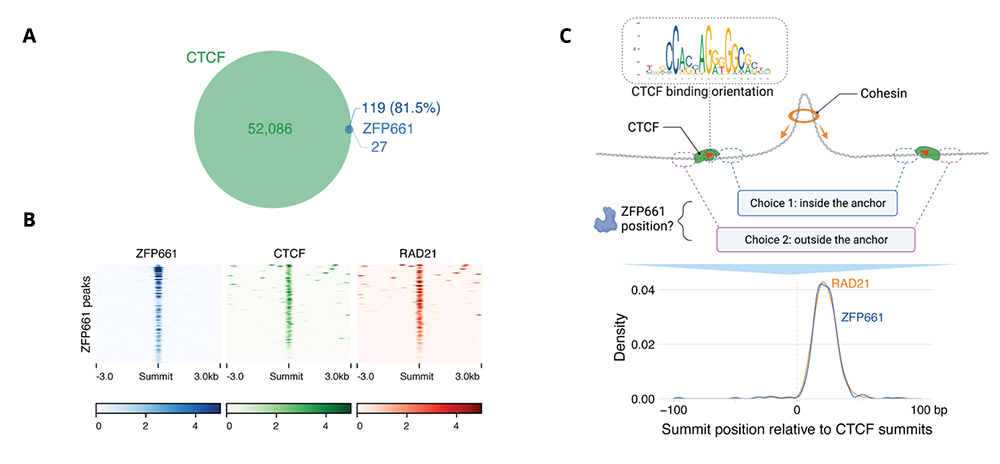
Figure 1. ZFP661 binds adjacent to a small subset of CTCF sites within loop anchors.
A. ZFP661 binding peaks overlap with a small subset of CTCF binding peaks.
B. Heat maps of ChIP-Seq signal across ZFP661 binding sites indicate strong overlap with CTCF and the cohesin subunit Rad21.
C. ZFP661 binding is found adjacent to CTCF binding motifs, within loop anchors, at the same position at which cohesin is typically trapped.
Dual histone methyl readers ZCWPW2 and ZCWPW1 connect PRDM9 to DNA double-strand breaks and their repair during meiotic recombination.
The dual histone-methylation writer PRDM9 is the most ancient KRAB–ZFP, which emerged in jawless fish and which plays a highly specialized role in meiotic recombination (MR). MR generates genetic diversity in sexually reproducing organisms and ensures proper synapsis and segregation of homologous chromosomes in gametes. Errors in MR that lead to mis-segregation of chromosomes are a leading cause of miscarriage and childhood disease. MR is initiated by programmed double-strand breaks (DSBs) in DNA that are distributed non-randomly at thousands of specific 1–2 kb regions called hotspots. In most mammals, hotspots are defined by PRDM9, a protein that contains a rapidly evolving DNA–binding zinc finger (ZF) array and a specialized HMT (histone methyltransferase) activity that catalyzes dual trimethylation marks on histone H3 at lysine 4 and lysine 36 (H3K4me3 and H3K36me3), both of whose activities are required for hotspot specification. Prdm9 loss of function causes sterility in mice, and PRDM9 mutations have been associated with male infertility in humans. In species lacking Prdm9, including yeast, plants, and birds, hotspots are located in H3K4me3–rich regions at gene promoters. Thus, the emergence of PRDM9 during evolution reshaped the MR landscape by relocating DSBs away from promoters to chromatin sites bound by the rapidly evolving PRDM9, which allowed for rapid interspecies hotspot diversification.
We set out to address whether other factors, in addition to PRDM9, are required to ‘re-engineer’ hotspot selection and how the DNA–break and –repair machinery is recruited to sites marked by PRDM9. We first identified the dual histone-methylation reader Zcwpw1, which co-evolved with and is tightly co-expressed with Prdm9. Using a mouse model, we found that ZCWPW1 is an essential meiotic recombination factor required for efficient repair of PRDM9–dependent DSBs and for pairing homologous chromosomes in male mice. However, ZCWPW1 is not required for the initiation of DSBs at PRDM9 binding sites. Our results indicate that the evolution of the dual histone-methylation writer (PRDM9) and reader (ZCWPW1) system in vertebrates remodeled genetic recombination–hotspot selection from an ancestral static pattern near genes towards a flexible pattern controlled by the rapidly evolving DNA–binding activity of PRDM9. Since publishing these findings, we identified a Zcwpw1 paralog, which was initially mis-annotated in the mouse genome, called Zcwpw2. Importantly we found that Zcwpw2 is essential for both mouse meiosis and fertility in males and females, and that it is important for the efficient generation of DSBs at hotspots relative to promoters. The studies thus revealed a three-component system, comprising the rapidly evolving DNA–binding histone methyltransferase (PRDM9) and the two dual histone-methylation readers (ZCWPW2 and ZCWPW1), which play at least partially separable roles in mediating the PRDM9–dependent generation of DNA DSBs and their repair at meiotic recombination hotspots.
Prdm9 KO mice are sterile, and PRDM9 variants/mutations have been associated with human infertility. PRDM9’s ZNF array is rapidly evolving (with a mutation rate about 100 times greater than a typical gene) leading to differential hotspot usage between individuals. Although humans and mice have more than 100 distinct PRDM9 alleles, the binding sites have only been mapped for a few variants (human A & C, mouse Dom2 & Cst). Thus, the drivers and biological consequences of PRDM9 allelic heterogeneity are unclear. We developed a cell-based high-throughput CUT&RUN assay to map where PRDM9 variants bind genome-wide using PRDM9–dependent H3K4me3 deposition as a proxy. We determined the binding sites and motifs of 89 previously uncharacterized human PRDM9 variants, including two novel/low-frequency variants we identified in a cohort of men with azoospermia (A7, C11). We found that, despite extensive ZNF composition differences, most human variants bound to known A or C hotspots, suggesting that many extant variants are functionally redundant. Some variants (n=8), including A7, bound to few locations (fewer than 5000) and had poorly defined motifs, suggesting they are nonfunctional. Other alleles (n=8), such as C11, bound to many more sites (over 30,000), most of which were distinct from the A/C variants. We mimicked the genotypes of the PRDM9-A/A7 and PRDM9-A/C11 in men with azoospermia by co-expressing both variants and found that A7 contributed minimally to PRDM9-A binding whereas C11 largely overrode it. Both allele types would likely reduce symmetric PRDM9 binding when paired with the major allele, leading to potential downstream negative impacts on gametogenesis and fertility. Our data provide a plausible mechanism for how PRDM9 zinc–finger array variation may contribute to human infertility.
To further explore a link between PRDM9 and infertility, we developed novel tools to genotype PRDM9 using long-read sequencing approaches, partnered with NICHD investigators studying infertility, and initiated an IRB–approved clinical study in collaboration with Shady Grove Fertility to genotype PRDM9 in a cohort of patients with non-obstructive azoospermia (NOA) and premature ovarian insufficiency (POI). In a preliminary analysis from 12 patients with POI, who had undergone genetic testing using conventional approaches where no causative gene could be identified, we identified both novel (n=1) as well as known but extremely low frequency alleles (n=3) of PRDM9. Using a combination of experimental and computational approaches, we demonstrated that these variants possess altered DNA–binding properties, supporting them as strong candidate genes in POI.
Additional Funding
- NIGMS Postdoctoral Research Associate Training Program Fellowship (Rachel Cosby)
Publications
- Young KRAB-zinc finger genes clusters are highly dynamic incubators of ERV-driven genetic heterogeneity in mice. Nat Commun 2025 16:9608
- The homeobox transcription factor MNX1 regulates the expression of many non-MN-specific neuronal genes in motor neurons. Nucleic Acids Res 2025 53:gkaf1015
- Meiosis specific distal cohesion site decoupled from the kinetochore. Nat Commun 2025 16:2116
- Patterns of recombination in snakes reveal a tug-of-war between PRDM9 and promoter-like features. Science 2024 383(6685):eadj7026
Collaborators
- Takashi Akera, PhD, Laboratory of Chromosome Dynamics and Evolution, NHLBI, Bethesda, MD
- Veronica Gomez-Lobo, MD, Pediatric Adolescent Gynecology, NICHD, Bethesda, MD
- Molly Przeworski, PhD, Columbia University, New York, NY
- Joana Vidigal, PhD, Laboratory of Biochemistry and Molecular Biology, Center for Cancer Research, NCI, Bethesda, MD
Contact
For more information, email todd.macfarlan@nih.gov or visit https://www.nichd.nih.gov/research/atNICHD/Investigators/macfarlan.