
Virulence Mechanisms of Microbial Pathogens

- Matthias Machner,
PhD, Chief, Section on Microbial Pathogenesis - Nicole Ellis, PhD, Staff Scientist
- Pablo Casino, PhD, Visiting Fellow
- Candice Chang, PhD, Visiting Fellow
- Ravi Pal, PhD, Visiting Fellow
- Marie Stoltzfus, PhD, Postdoctoral Fellow
- Emma Hunter, BSc, Postbaccalaureate Intramural Research Training Award Fellow
- Aisling Macaraeg, BSc, Postbaccalaureate Intramural Research Training Award Fellow
- Emma Neiswender, BSc, Postbaccalaureate Intramural Research Training Award Fellow
Our main research goal is to study how pathogenic bacteria subvert the human host defense and cause disease. In parallel, we investigate whether such mechanisms may be manipulated for preventative and/or therapeutic purposes. As a model organism, we use the bacterium Legionella pneumophila, the causative agent of a potentially fatal respiratory infection known as Legionnaires' disease. According to the CDC (Centers for Disease Control), the number of Legionnaires' disease cases in the U.S. has risen more than four-fold over the past 20 years, making L. pneumophila an emerging pathogen of increasing relevance. Contrary to what its name may imply, Legionnaires’ disease occurs in individuals of all ages, including children who receive respiratory therapy, newborns who had recently undergone surgery or under-water birth, and children who are immune-compromised. We are committed to an in-depth analysis of the mechanisms that allow L. pneumophila to exploit the human host and cause disease. Insights gained from our studies will ultimately improve our ability to diagnose, prevent, and fight Legionnaires’ disease and related illnesses, thereby contributing to the success of NICHD’s mission.
Within freshwater environments, L. pneumophila exists as an intracellular parasite of single-cell organisms known as amoeba. Upon inhalation of contaminated water droplets, L. pneumophila enters the lung and is phagocytosed (taken up) by specialized immune cells known as alveolar macrophages (Figure 1). Instead of being degraded by these cells, the pathogen establishes a protective membrane compartment, the Legionella-containing vacuole (LCV). Within this intravacuolar niche, L. pneumophila can replicate to high numbers before killing the host cell and infecting neighboring cells.
Figure 1. Intracellular replication cycle of Legionella pneumophila
Upon uptake by a macrophage, L. pneumophila delivers a large number of effector proteins (red) through the Dot/Icm type IV secretion system (T4SS) into the host cytosol. The effectors manipulate signaling and trafficking pathways in order to establish conditions favorable for L. pneumophila growth. Eventually, the host cell is lyzed, and L. pneumophila bacteria infect neighboring cells.
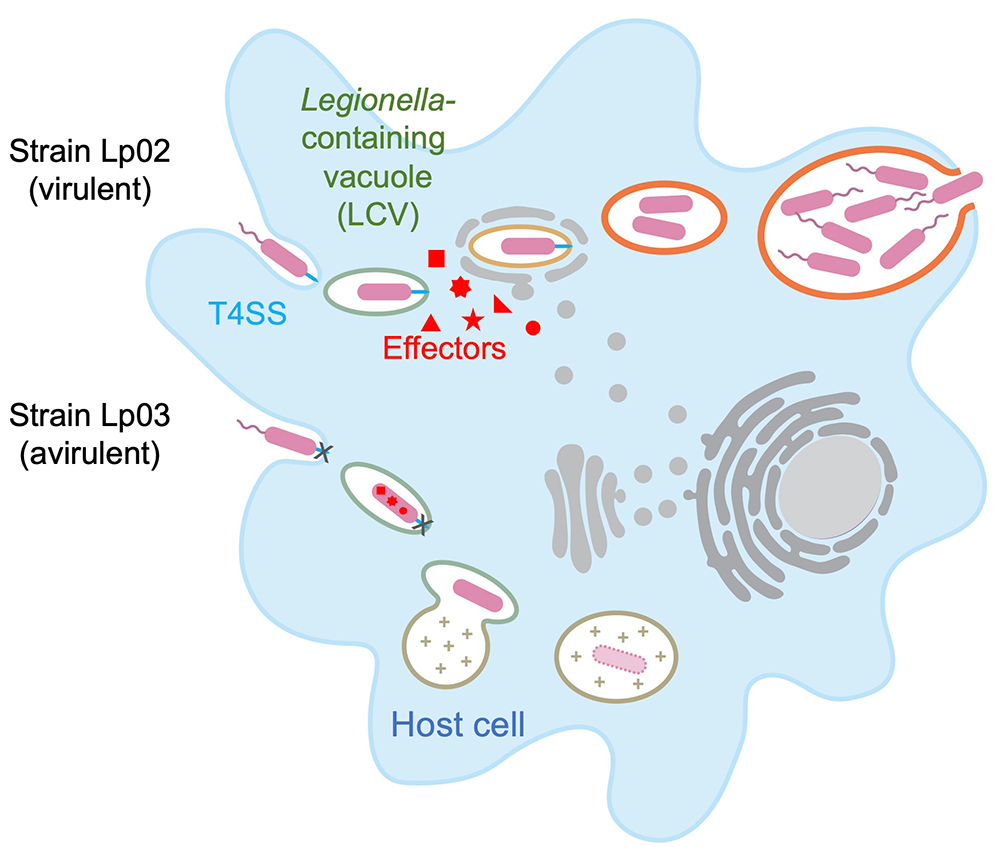
Figure 1. Intracellular replication cycle of Legionella pneumophila
Upon uptake by a macrophage, L. pneumophila delivers a large number of effector proteins (red) through the Dot/Icm type IV secretion system (T4SS) into the host cytosol. The effectors manipulate signaling and trafficking pathways in order to establish conditions favorable for L. pneumophila growth. Eventually, the host cell is lyzed, and L. pneumophila bacteria infect neighboring cells.
The virulence of L. pneumophila relies on the activity of more than 300 proteins, or effectors, that are delivered into the host cytosol by a specialized translocation apparatus called the Dot/Icm type IV secretion system (T4SS). L. pneumophila mutants with a non-functional T4SS are degraded by macrophages (Figure 1), underscoring the importance of the translocated effectors for host-cell manipulation and bacterial virulence.
Our main objective is to obtain detailed mechanistic insight into the L. pneumophila virulence program by investigating the biological role of its effectors at a molecular, cellular, and structural level. Knowledge obtained from these studies can help in the development of novel therapeutics aimed at treating or preventing Legionnaires' disease and related illnesses.
Identification of virulence-critical genes using CRISPR interference
A major obstacle in deciphering Legionella virulence mechanisms is that the deletion of effector-encoding genes, individually or even in groups, rarely results in detectable intracellular growth defects, a phenomenon that is attributed to the existence of a significant level of overlap in the effector's function and/or host targets, so that the loss of individual effectors is compensated for by the activity of synergistic or redundant effectors. Classical genetic approaches such as transposon mutagenesis or chemical mutagenesis, which target genes at random and often disrupt house-keeping genes essential for viability, have been mostly unsuccessful in identifying combinations of virulence-critical genes of Legionella, other than those encoding components of the essential Dot/Icm apparatus.
We created a novel gene-silencing tool in Legionella that harnesses the power of CRISPR interference (CRISPRi) to silence pairs or even entire groups of bacterial genes so as to systematically screen for combinations of virulence-critical effectors in this pathogen (Figure 2). CRISPRi requires the presence of three functional components: a protein called dCas9, which is the nuclease-deficient form of the Streptococcus pyogenes Cas9 enzyme; a trans-activating CRISPR (tracr) RNA that interacts with both dCas9; and the third component of this system, the CRISPR (cr)RNA. Together with the tracrRNA, crRNAs specifically direct dCas9 through base-pairing to complementary target DNAs, thus imposing a road block that prevents gene expression by sterically precluding RNA polymerase from gaining access to the gene (Figure 2). In nature, CRISPR/Cas systems are found in the majority of bacterial and archaeal genomes, where they serve as a naturally occurring adaptive immune system. Fragments of foreign DNA, such as virus DNA, transposable elements, or plasmids, are degraded, and fragments are incorporated into the bacterial chromosome as a series of spacers separated by repeats. Such repeat-spacer-repeat arrays, which can contain dozens or even hundreds of unique spacers, serve as a memory of past infections and protect the bacteria from re-infection.
Figure 2. Concept of CRISPRi
The three components dCas9 (blue), tracrRNA (black), and crRNA (red) form a trimeric complex that binds to target genes via homologous base pairing and blocks transcription by RNA polymerase. Ptet, promoter sequence.
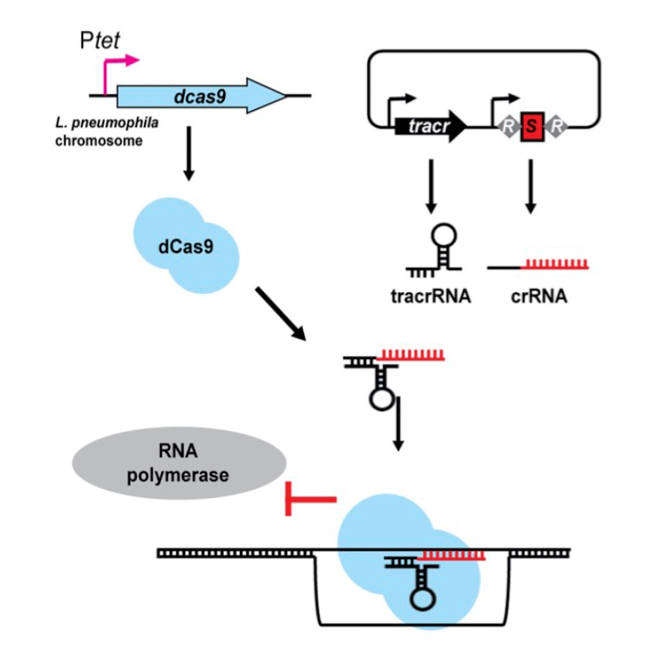
Figure 2. Concept of CRISPRi
The three components dCas9 (blue), tracrRNA (black), and crRNA (red) form a trimeric complex that binds to target genes via homologous base pairing and blocks transcription by RNA polymerase. Ptet, promoter sequence.
Given the natural multiplex (meaning multiple gene targets at a time) capability of CRISPRi, we reasoned that this system could be exploited in Legionella to simultaneously silence entire families of chromosomal genes simply by replacing the foreign DNA spacer sequences of CRISPR arrays with sequences complementary to the bacterium’s own genes (Figure 2). The dCas9–encoding gene was integrated into the Legionella genome, while the tracrRNA– and crRNA–encoding sequences were provided on a plasmid, each under control of the anhydrous tetracycline-inducible tet promoter. Using biochemical and cell-based assays, we confirmed that our CRISPRi approach was tunable, specific, and functional during intracellular growth. Most importantly, it was multiplex-capable, allowing us to silence up to ten genes at a time in Legionella. Upon interrogation of nearly 200 effector-encoding genes using synthesized arrays of ten repeat-spacer-repeat elements, we discovered several combinations of genes that, when silenced simultaneously, led to profound growth defects of Legionella in human macrophages or amoeba, notably without compromising bacterial growth outside the host. Such newly discovered virulence genes represent promising new targets for therapeutic intervention.
Design of a next-generation randomized multiplex CRISPRi approach
A major limitation of the proof-of-concept CRISPRi approach described above was that it was not randomized, given that the 10-plex CRISPR arrays had to be commercially synthesized, meaning that they each targeted a predetermined group of ten specific Legionella genes. To bypass this limitation, we generated an improved CRISPRi tool, whereby the CRISPR arrays were self-assembled in a randomized (rather than predetermined) fashion from oligonucleotide pairs. We succeeded in assembling a collection of thousands of plasmids containing hundreds of unique combinations of arrays able to silence between two and eleven genes (average of 3.1 genes), serving as the foundation for the first multiplex, randomized CRISPR interference sequencing (MuRCiS) pipeline (Figure 3). We also designed a customized bioinformatics algorithm that monitored differences in array abundance in the output (post infection) vs. input pool, where a reduction in the abundance of an array was indicative that it had silenced gene combinations indispensable for intracellular replication.
Figure 3. Schematic overview of the experimental and bioinformatics pipeline of MuRCiS
During MuRCiS, a pooled population of L. pneumophila (dcas9) strains bearing the multiplex random CRISPR arrays were subjected to a selective pressure (intracellularity). Plasmids were purified from both input and output bacteria populations, linearized, and submitted for long-read PacBio Sequel sequencing. For simplification, an array of mixed spacer population is shown as a single stretch of color, each color representing a different combination of spacers. In this example, the yellow and red arrays are lost in the output, suggesting that they silence critical combinations of genes.
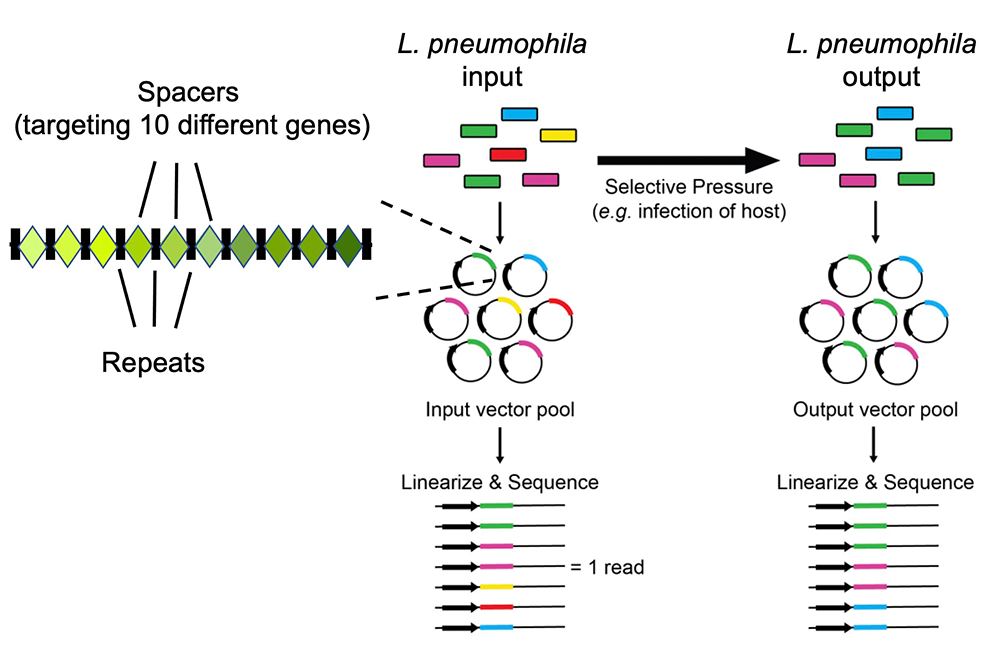
Figure 3. Schematic overview of the experimental and bioinformatics pipeline of MuRCiS
During MuRCiS, a pooled population of L. pneumophila (dcas9) strains bearing the multiplex random CRISPR arrays were subjected to a selective pressure (intracellularity). Plasmids were purified from both input and output bacteria populations, linearized, and submitted for long-read PacBio Sequel sequencing. For simplification, an array of mixed spacer population is shown as a single stretch of color, each color representing a different combination of spacers. In this example, the yellow and red arrays are lost in the output, suggesting that they silence critical combinations of genes.
In a proof-of-concept study, we used MuRCiS to probe 44 highly conserved transmembrane domain (TMD)–containing effectors of Legionella for their importance during bacterial replication in both U937 macrophages (the disease host) and Acanthamoeba castellanii (the environmental host), and several gene combinations that were identified as vital have become the focus of our future research. The MuRCiS platform is applicable to other groups of gene families besides T4SS effectors and will undoubtedly identify more interesting biology related to Legionella pathogenesis.
The Legionella pneumophila effector DenR hijacks the host NRas proto-oncoprotein to downregulate MAPK signaling.
While studying intracellular trafficking of Legionella in human HT1080 cells, we discovered that the host protein N-Ras accumulates on LCVs (Figure 4). This was an unusual finding, given that Ras proteins, unlike GTPases of the Rab, Arf, Rho, and Ran subfamily, have not been shown to be targeted by Legionella. N-Ras is a member of the Ras subfamily of small GTPases. It transmits extracellular inputs from the plasma membrane (PM) along various signaling pathways to promote cell growth, proliferation, and survival. If mutated, Ras GTPases are responsible for 30% of all human cancers, with N-Ras mutations occurring in 15–20% of all melanomas.
Figure 4. Model explaining how NRas recruitment dampens MAPK signaling from the PM while initiating alternative signaling from the LCV
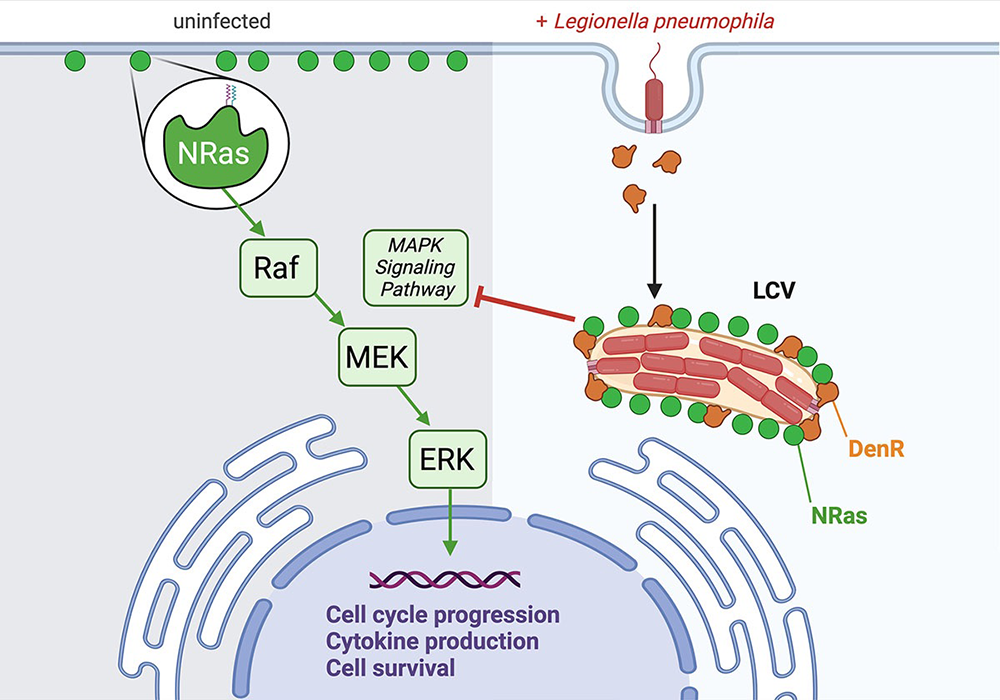
Figure 4. Model explaining how NRas recruitment dampens MAPK signaling from the PM while initiating alternative signaling from the LCV
We used site-directed mutagenesis to show that the two sites for lipidation, specifically farnesylation and S-acylation, within the C-terminal hypervariable region (HVR) of N-Ras are required for Ras trafficking to the LCV. Using our multiplex CRISPRi tool described above, we found that silencing of a single gene, called denR, rendered Legionella defective for N-Ras recruitment (hence, denR). A strain with an in-frame deletion in denR (Lp02DdenR) phenocopied this recruitment defect and could be complemented with plasmid-encoded DenR. Using a translational beta-lactamase-reporter fusion, we verified that DenR is a bona fide T4SS effector. Co-precipitation assays from lysate of transiently transfected cells producing GFP–DenR and Halo–tagged N-Ras confirmed that the two proteins exist in a complex within eukaryotic cells. Furthermore, upon production of GFP–DenR in transiently transfected HT1080 cells, we confirmed that DenR specifically localized to LCVs, as is to be expected for an effector that recruits N-Ras during infection.
To examine how redirecting N-Ras away from the PM and to the LCV affected cellular signaling, we performed comparative label-free global proteomics and phospho-proteomics on lysate collected from RAW264.7 macrophages challenged with either Lp02ΔdenR or the complemented strain Lp02ΔdenR(pdenR). We identified DenR–mediated alterations in several pathways known to be associated with Ras downstream signaling, most notably the well characterized mitogen-activated protein kinase (MAPK) signaling pathway (Figure 4). Using phospho-specific antibodies, we confirmed a reduction in the levels of activated phospho–MEK (MAPK/ERK kinase) and phospho–ERK (extracellular-signal regulated kinase) concomitant with increasing amounts of DenR.
In summary, our data provide evidence for a previously unrecognized involvement of Ras subfamily GTPases in Legionella pathogenesis, where DenR, by redirecting N-Ras away from the PM and to the LCV, dampens signaling cascades originating from the PM, most notably the MAPK pathway (Figure 4).
Additional Funding
- NICHD Early Career Awards FY25 (to Ravi Pal)
Publications
- The Legionella pneumophila effector DenR hijacks the host NRas proto-oncoprotein to downregulate MAPK signaling. Cell Rep 2024 43:114033
- A randomized multiplex CRISPRi-Seq approach for the identification of critical combinations of genes. Elife 2023 12:RP86903
- A multiplex CRISPR interference tool for virulence gene interrogation in Legionella pneumophila. Commun Biol 2021 4:157
- Genetic approaches for identifying and characterizing effectors in bacterial pathogens. Annu Rev Genet 2024 58:233-247
Collaborators
- Timothy J. Donohue, PhD, Great Lakes Bioenergy Research Center, University of Wisconsin-Madison, Madison, WI
- Aravind Iyer, PhD, Protein and Genome Evolution Research Group, NLM/NCBI, Bethesda, MD
- Michal Jarnik, PhD, Section on Intracellular Protein Trafficking, NICHD, Bethesda, MD
- Doreen Matthies, PhD, Unit on Structural Biology, NICHD, Bethesda, MD
- Kevin S. Myers, PhD, Great Lakes Bioenergy Research Center, University of Wisconsin-Madison, Madison, WI
- Aleksandra Nita-Lazar, PhD, Functional Cellular Networks Section, NIAID, Bethesda, MD
- Dhirendra Simanshu, PhD, Structural Biology, NCI RAS Initiative at Frederick National Laboratory for Cancer Research, Frederick, MD
- Alexander J. Sodt, PhD, Unit on Membrane Chemical Physics, NICHD, Bethesda
- Chad Williamson, PhD, Advanced Microscope Facility, NICHD, Bethesda, MD
Contact
For more information, email machnerm@mail.nih.gov or visit https://www.nichd.nih.gov/research/atNICHD/Investigators/machner.