
Pathophysiology, Genetics, and Treatment of Congenital Adrenal Disorders

- Deborah P. Merke,
MD, MS, Adjunct Investigator, Chief, Section of Congenital Disorders, Clinical Center - Vipula Kolli, PhD, Staff Scientist
- Qizong Lao, PhD, Staff Scientist
- Sonal Vaid, MD, Staff Clinician
- Elizabeth Joyal, MSN, Nurse Practitioner
- Peyton Lee, BA, Postbaccalaureate Intramural Research Training Award Fellow
- Molly Ruebusch, BS, Postbaccalaureate Intramural Research Training Award Fellow
- Jacob Trick, BS, Postbaccalaureate Intramural Research Training Award Fellow
- Christina Tatsi, MD, PhD, Physician, Contractor
In its most severe classic form, congenital adrenal hyperplasia (CAH) is a life-threatening, rare orphan disease that is part of the neonatal screen performed in all 50 U.S. states. In its mildest non-classic form, CAH is one of the most common autosomal recessive diseases and may be a common cause of female infertility. Our research program strives to elucidate the pathophysiology and genetics of CAH, thus facilitating the development of new approaches to the diagnosis, evaluation, and treatment of the disease. We are conducting the largest ever Natural History Study of CAH, with over 450 patients enrolled. We were the first to identify adrenaline deficiency as a new hormonal imbalance in CAH and the first to report smaller-than-normal amygdala, the emotion regulator of the brain, in CAH, providing insight into hormonal effects on the brain. We also found that approximately 15 percent of patients with CAH owing to 21-hydroxylase deficiency have a contiguous gene-deletion syndrome resulting in connective tissue dysplasia and a hypermobility-type Ehlers-Danlos syndrome, which represents a novel phenotype and which we named CAH-X. Our studies over the years have had a major impact on the management of patients with CAH, including changing the recommended stress-dosing practices in the Endocrine Society Clinical Practice Guidelines and, more recently, establishing recommendations for the management of women with CAH during pre-conception, pregnancy, and postpartum, and the development of a disease-specific patient-reported outcome instrument to assess health-related quality of life. Central to our work is the study of new treatments, including a long-term trial testing sex-hormone blockade in children, novel ways of replacing cortisol aimed at mimicking the normal circadian rhythm of cortisol secretion, antagonists of the CRH (corticotropin-releasing hormone) type 1 receptor to suppress the drivers of excess androgen production so as to allow for lower-dose glucocorticoid therapy, and gene therapy. The NIH Clinical Center is the ideal venue in which to carry out such studies and is one of the few places in the world that facilitates the conduct of long-term studies of rare diseases. Through the study of a large cohort of these unique patients, there is great potential to expand our understanding of hormonal effects on normal development and physiology, to provide insights into gene regulation and expression, and to develop and test new treatment approaches.
Genetic studies
CAH is most commonly caused by 21-hydroxylase deficiency. The gene encoding 21-hydroxylase, CYP21A2, and a highly homologous pseudogene, CYP21A1P, map to the short arm of chromosome 6 within the human leukocyte antigen histocompatibility complex. The deleterious sequence in the CYP21A1P pseudogene can be transferred to the CYP21A2 functional gene by homologous recombination, and such events produce common mutations, which account for approximately 95% of all CYP21A2 disease–causing mutations. Of the common mutations, approximately 30% are large deletions. The TNXB gene, encoding tenascin-X, an extracellular matrix protein that is highly expressed in connective tissue, and TNXA, a highly homologous pseudogene, flank CYP21A2 and CYP21A1P, respectively. Autosomal recessive tenascin-X deficiency was described as a cause of Ehlers-Danlos syndrome in 2001. We hypothesized that deletions of CYP21A2 might commonly extend into the TNXB gene, and we have been studying this phenomenon in our Natural History Study.
The first evaluation of the potential clinical implications of TNXB heterozygosity in CAH patients was performed in our Natural History Study of CAH (www.ClinicalTrials.gov Identifier No. NCT00250159) at the NIH Clinical Center. In 2013, we prospectively studied 193 consecutive, unrelated patients with CAH with clinical evaluations for manifestations of Ehlers-Danlos syndrome and genetic evaluations for TNXB mutations. Heterozygosity for a TNXB deletion was present in 7% of CAH patients; such CAH patients were more likely than age-and sex-matched CAH patients with normal TNXB to have joint hypermobility, chronic joint pain, multiple joint dislocations, and a structural cardiac valve abnormality detected by echocardiography. Six of 13 probands had a cardiac abnormality, including the rare quadricuspid aortic valve, a left ventricular diverticulum, and an elongated anterior mitral valve leaflet. As a result of the study, the term CAH-X was coined to describe the subset of CAH patients who display an Ehlers-Danlos syndrome phenotype resulting from the monoallelic presence of a CYP21A2 deletion extending into the TNXB gene.
The study of CAH-X has provided insight into the recombination events that occur in the class III region of the major histocompatibility complex (MHC) locus, a region in the genome that is predisposed to genetic recombination and misalignment during meiosis. The majority of deletions generate chimeric CYP21A1P/CYP21A2 genes (Figure 1). Chimeric recombination between TNXB and TNXA also occurs (Figure 1). The recombination event deletes CYP21A2 and therefore represents a CAH disease–causing allele. We described three unique types of TNXA/TNXB chimera (CH): CAH-X CH-1 renders the gene nonfunctional, resulting in reduced dermal and serum TNX expression; CAH-X CH-2 alters protein structure; and CAH-X CH-3 is predicted to reduce protein-folding energy. Our lab continues to investigate how TNXB contributes to the phenotype of CAH patients, and to identify novel chimeric genes.
Figure 1. Schematic of CYP21A1P/CYP21A2 and TNXA/TNXB chimeric genes
Formation of chimeric genes occurs as a result of misalignment of homologous genes during meiosis. Active genes are in solid colors; pseudogenes are in grey and are framed with the color of the corresponding functional gene. Representative chimeric genes are shown. In total, there are nine known CYP21A1P/CYP21A2 chimeras (CH-1 to CH-9), and we identified three different types of TNXA/TNXB chimeras (CAH-X CH-1 to CAH-X CH-3) with different junction sites. Approximately 15 percent of patients with CAH owing to 21-hydroxylase deficiency carry at least one TNXA/TNXB chimera, resulting in hypermobility-type Ehlers-Danlos syndrome or CAH-X syndrome.
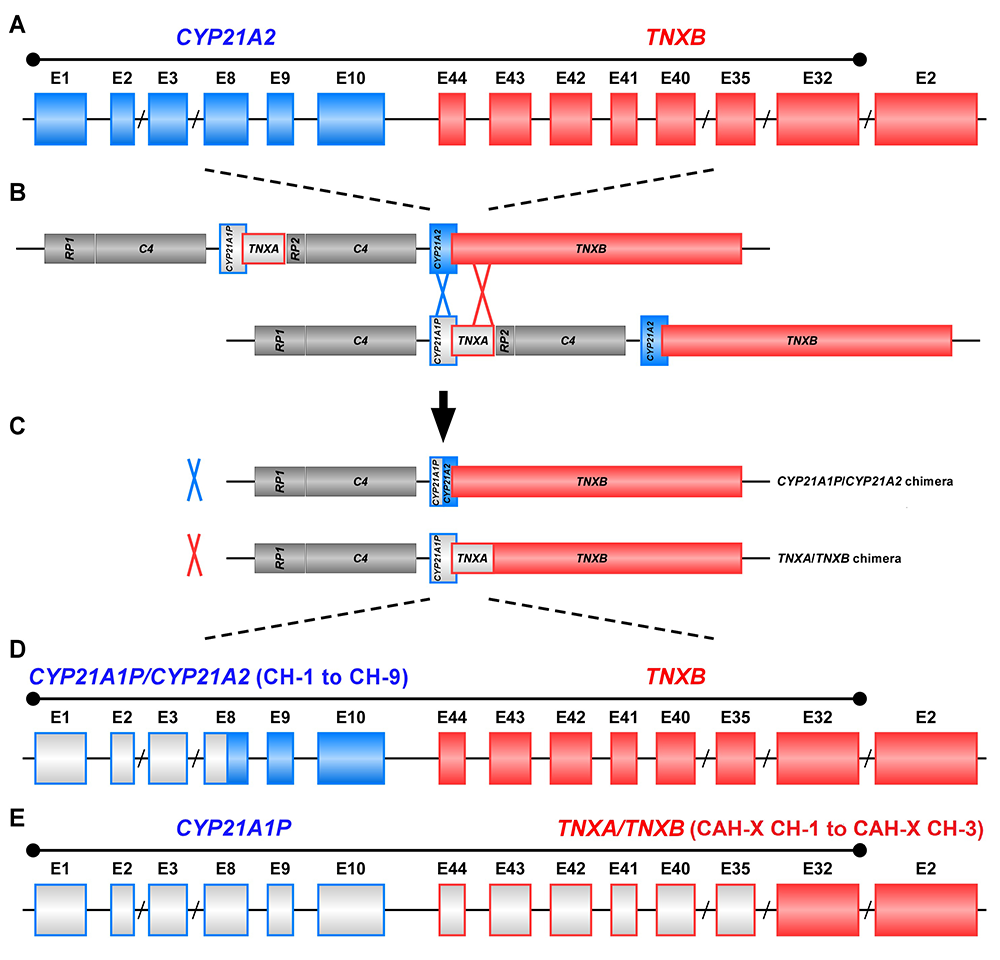
Figure 1. Schematic of CYP21A1P/CYP21A2 and TNXA/TNXB chimeric genes
Formation of chimeric genes occurs as a result of misalignment of homologous genes during meiosis. Active genes are in solid colors; pseudogenes are in grey and are framed with the color of the corresponding functional gene. Representative chimeric genes are shown. In total, there are nine known CYP21A1P/CYP21A2 chimeras (CH-1 to CH-9), and we identified three different types of TNXA/TNXB chimeras (CAH-X CH-1 to CAH-X CH-3) with different junction sites. Approximately 15 percent of patients with CAH owing to 21-hydroxylase deficiency carry at least one TNXA/TNXB chimera, resulting in hypermobility-type Ehlers-Danlos syndrome or CAH-X syndrome.
It is now estimated that approximately 15% of patients with CAH resulting from 21-hydroxylase deficiency are affected by CAH-X. Overall, CAH-X patients have generalized joint hypermobility, sub-luxations, and chronic arthralgia, and about 25% have cardiac structural abnormalities. Patients with biallelic CAH-X show severe skin hyperextensibility, with delayed wound healing and significant joint hypermobility. Other connective-tissue disease manifestations in CAH-X patients include chronic tendonitis and/or bursitis, rectal prolapse, severe gastroesophageal reflux, and cardiac abnormalities. Genetic testing for CAH-X is complex and complicated by pseudogene interference and the large (70kb) size of the TNXB gene.
This year, we reported the clinical and biochemical phenotype across the genotypic spectrum of CAH due to 21-hydroxylase deficiency in a cohort of 457 patients. We found that 0.33% of pathogenic variants arose de novo, and 10.1% had CAH-X. CAH is diagnosed based on levels of 17-hydroxprogesterone (17OHP), and we found that levels of 17OHP at diagnosis varied significantly by five genotype groups; but maximum values obtained during clinical care over time were similar among all classic (severe form) and among all non-classic (mild form) genotypes. The variant with a P30L mutation is expected to have a non-classic phenotype; however, patients often have more severe symptomatology. Patients with the P30L variant had significantly higher 17OHP and lower cortisol at diagnosis compared with other NC genotypes. Our data supports the consideration of P30L genotype as a unique group. Our findings are important to guide genetic counseling and provide anticipatory guidance in disease management.
We continue to refine and improve genetic testing methodology in our research.
Development of a patient-reported outcome instrument to assess health-related quality of life in congenital adrenal hyperplasia
Measuring health-related quality of life (HRQoL) is a crucial and often required process for evaluating health care outcomes. For chronic medical conditions such as congenital adrenal hyperplasia (CAH), for which a cure does not exist, HRQoL instruments provide insight into the fluctuations in disease severity and the effectiveness of longitudinal clinical management. HRQoL instruments have been successfully developed for other endocrine and chronic conditions.
Classic CAH affects 1 in 10,000 to 1 in 20,000 live births. It is a complex, chronic genetic condition with multiple hormonal imbalances, and the adverse outcomes are the result of disease- and treatment-related factors, including adrenal insufficiency, androgen excess, and adverse effects of chronic exogenous glucocorticoid excess. Several disease-related comorbidities, reduced HRQoL, and increased mortality has been reported in this patient population (Figure 2).
Figure 2. Clinical features of congenital adrenal hyperplasia
Disease-related manifestations are shown in blue, treatment-related manifestations are shown in red, and clinical manifestations related to both the disease and the treatment are shown in brown.
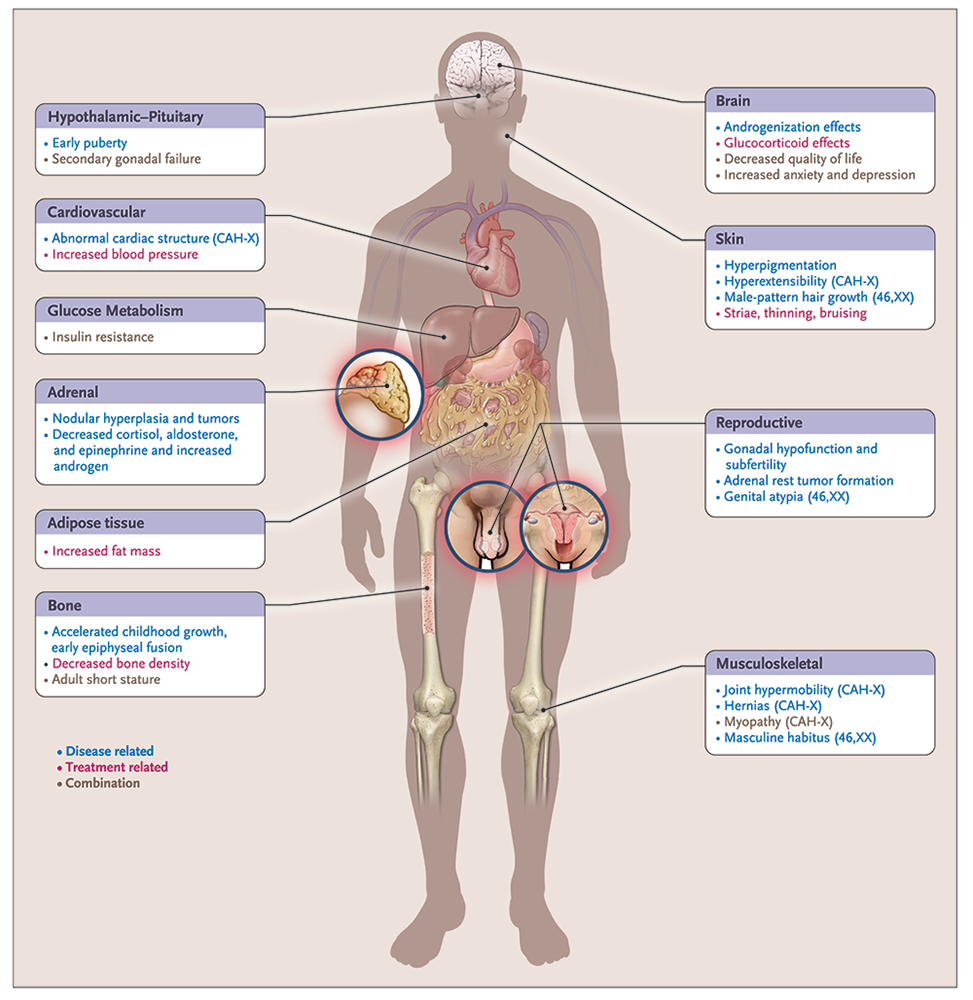
Figure 2. Clinical features of congenital adrenal hyperplasia
Disease-related manifestations are shown in blue, treatment-related manifestations are shown in red, and clinical manifestations related to both the disease and the treatment are shown in brown.
Our long-term natural history study has been critical for understanding disease-specific outcomes, and has enabled us to develop a CAH–specific patient-reported quality-of-life questionnaire (CAHQL) for adolescents and adults that captures the unique challenges that CAH imposes on the well-being of the affected population.
The CAHQL was developed following the Food and Drug Administration (FDA) recommendations for the construction of patient-reported outcome measures and the end-point model for reporting outcomes of clinical trials. The first step involved review of the literature, obtaining expert input, and interviewing a representative sample of the target population, which was used to develop a conceptual framework for the instrument. Open-ended questions and semi-structured questions about issues anticipated to be relevant to the patient were asked. Parents were interviewed to obtain additional insight into the impact of CAH on the individual and the family. Content validity was established with performance of cognitive interviews of patients and experts. A recall period of four weeks was deemed appropriate for collection of HRQoL data. Internal consistency of the defined domains was evaluated using Cronbach’s alpha, a reliability coefficient and a measure of the internal consistency of tests and measures. For items that showed poor inter-domain consistency, exploratory factor analysis was used to determine whether there was a better fit in a different domain. The final CAHQL questionnaire has 44 items and 7 domains (Figure 3).
Figure 3. Flowchart of the development of a patient-reported outcome instrument (CAHQL) to assess health-related quality of life in congenital adrenal hyperplasia
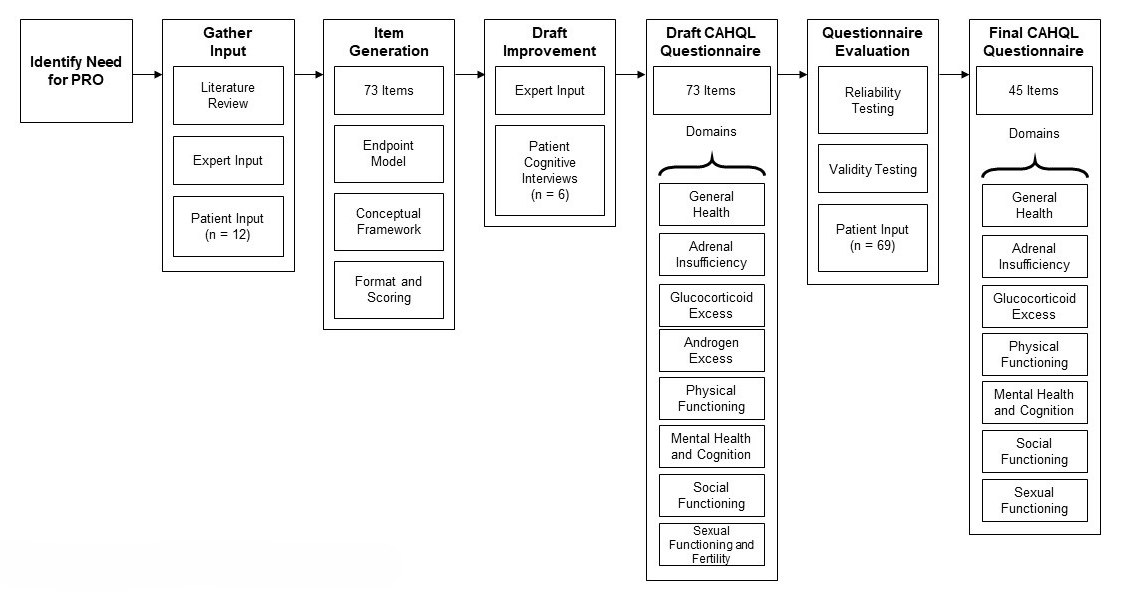
Figure 3. Flowchart of the development of a patient-reported outcome instrument (CAHQL) to assess health-related quality of life in congenital adrenal hyperplasia
Classic CAH is unique in its physiology; patients struggle with symptoms of adrenal insufficiency, fear of an adrenal crisis, and risk for hospitalization, while experiencing the long-term effect of chronic supra-physiological glucocorticoid exposure, often needed to control corticotropin-driven excess androgens. Adverse outcomes may occur throughout life as the result of multiple hormonal imbalances. The final CAHQL questionnaire that we developed is the first disease-specific instrument to assess the multidimensional HRQoL of patients with classic CAH and to capture all pertinent aspects of classic CAH on patient well-being, involving both physical and mental functioning. In addition to its anticipated use in the clinical setting, the instrument may be used to assess the efficacy of novel treatments in development.
Novel treatment approaches: hypothalamic-pituitary-adrenal axis suppression
Excess glucocorticoid therapy is often needed to control the overproduction of adrenal androgens in CAH. Potential strategies to address the drivers of excess androgen production in CAH include suppression of the hypothalamic-pituitary-adrenal axis. High-affinity and selective small-molecule antagonists of corticotropin-releasing factor type 1 (CRF1) receptors in the pituitary gland were developed to potentially allow for lower glucocorticoid therapy. The mechanism of action was validated in a previous proof-of-concept study and was shown to be safe in Phase 1 clinical studies in healthy volunteers and Phase 2 dose-finding studies of patients with CAH.
A small-molecule antagonist of the CRF1 receptor antagonist (crinecerfont) was studied in a multicenter, randomized, double-blind, placebo-controlled, 6-month clinical trial in adults with CAH (NCT04490915) to investigate the effect of CRF1 antagonist on outcomes such as improved adrenal androgen control, as well as the potential for reducing glucocorticoid dose as an add-on therapy. The mean glucocorticoid dose at baseline was 17.6 mg per square meter of body-surface area per day of hydrocortisone equivalents; the mean androstenedione level was elevated at 620 ng per deciliter. At week 24, the change in the glucocorticoid dose (with androstenedione control) was −27.3% in the CRF1 receptor–antagonist group and −10.3% in the placebo group (least-squares mean difference, −17.0 percentage points). A physiologic glucocorticoid dose (with androstenedione control) was reported in 63% of the patients in the CRF1 receptor antagonist group and in 18% in the placebo group. The use of a CRF1 receptor antagonist resulted in a significantly greater decrease from baseline in the mean daily glucocorticoid dose, including a reduction to the physiologic range, than placebo, following evaluation of adrenal androgen levels. This alternative strategy for controlling excess adrenal androgens enables reduction of glucocorticoid doses to a physiologic or near-physiologic range with reduced adrenal androgen production. In December 2024, The FDA approved the use of crinecerfont as an adjunctive treatment to glucocorticoid replacement to control adrenal androgens in adults and pediatric patients four years of age and older with classic CAH. Long-term outcomes are needed.
Novel treatment approaches: sex steroid blockade and inhibition
As an alternative approach to the treatment of CAH, the effects of elevated androgen and estrogen could be prevented through the use of sex-steroid blockade. Short-term (two-year) administration of an antiandrogen and aromatase inhibitor and reduced hydrocortisone was shown to normalize linear growth rate and bone maturation. In 2025, a prospective long-term, randomized, parallel study to adult height of an antiandrogen (flutamide) and an aromatase inhibitor (letrozole), and reduced hydrocortisone dose vs. standard treatment of 62 children with classic CAH was completed (NCT00001521).
Although the primary endpoint, adult height, did not differ between the two treatment groups (conventional therapy vs. combination regimen of antiandrogen, aromatase inhibitor, and reduced hydrocortisone and fludrocortisone), several lessons were learned. First, obtaining adult height outcomes is essential when evaluating potential height-enhancing therapy in children. Short-term therapies that improve growth and predicted adult height in growing children do not necessarily translate to improved adult height. In this long-term study to adult height, significantly greater improvement in predicted adult height was observed at pubertal onset in the investigational arm compared with conventional therapy. Second, a physiological daily dose of hydrocortisone of approximately 8 mg/m2day can be safely given to children. The incidence of adrenal crises was similar to prior population-based studies and not increased. This finding is important, as newer adjuvant medications in development aim to allow for lower hydrocortisone dosing. Third, although pre-pubertal cortisol and sex-steroid exposures may influence the growth plate and hence growth potential, hormonal exposures during puberty have a major influence on adult height. Lastly, our data do not support the use of aromatase inhibitors in children to improve adult height outcome.
The ultimate determinant of whether or not prepubertal treatments are effective in improving the growth of children is adult height. The Clinical Center is the ideal place to carry out such a long-term study.
Novel treatment approaches: gene therapy for congenital adrenal hyperplasia through administration of an adeno-associated virus (AAV) serotype 5–based recombinant vector encoding the human CYP21A2
Classic CAH remains life threatening and difficult to manage with current treatment options, highlighting the need for effective therapies that can have appreciable long-term impacts. For example, adrenal crisis can rapidly lead to severe shock and death even in patients who are compliant with therapy. Significantly, the risk for all-cause mortality rates in classic CAH patients was over five times that of matched controls, adjusting for age and sex. In addition, and despite current standard of care, classic CAH patients continue to have an increased incidence of cardiovascular and metabolic disorders, reduced lifespan, and reduced quality of life.
Gene therapy for CAH resulting from 21-hydroxylase deficiency was first studied 20 years ago, when an intra-adrenal injection of an adenoviral vector transiently restored enzyme activity in 21-hydroxylase–deficient mice for a few days. Advances in adeno-associated virus (AAV) vector–based therapies have expanded the gene therapy landscape to include CAH. An intravenous injection of an AAV–CYP21 vector restored steroidogenesis and demonstrated phenotypic changes (weight gain) in mice for more than 15 weeks. Subsequent non-human primate studies demonstrated durable expression of vector genome copies. Preclinical animal studies examining efficacy, safety, and biodistribution supported clinical development of the AAV5 vector, and a first-in-human study was conducted in adults, designed for assessment of safety and efficacy (NCT04783181). At higher doses, some efficacy was observed consistent with transgene deliver to the adrenals; however, the activity generated was insufficient to rescue adrenal insufficiency. Further human dosing in this study has been suspended.
Additional Funding
- Cooperative Research and Development Agreement (CRADA) for: Age-Appropriate Hydrocortisone Formulations for the Treatment of Adrenal Insufficiency including Congenital Adrenal Hyperplasia
- Cooperative Research and Development (CRADA) for: Randomized, Double-Blind, Placebo-Controlled Study to Evaluate the Safety and Efficacy of Crinecerfont in Adult Subjects with CAH, Followed by Open-Label Treatment
- Cooperative Research and Development Agreement (CRADA) for: Study of the Safety and Pharmacodynamic Activity of Gene Therapy for Congenital Adrenal Hyperplasia
Publications
- CAHQL: A patient reported outcome instrument to assess health-related quality of life in congenital adrenal hyperplasia. J Clin Endocrinol Metab 2025 110(4):e1149-e1159
- Clinical and biochemical phenotype across the genotypic spectrum of 21-hydroxylase deficiency in 457 individuals. J Clin Endocrinol Metab 2025 Online ahead of print
- Adult height following prepubertal treatment with an antiandrogen, aromatase inhibitor and reduced hydrocortisone in children with classic congenital adrenal hyperplasia. J Clin Endocrinol Metab 2025 110(7):e2171-e2182
- Longitudinal characterization and sonographic staging of testicular adrenal rest tumors. J Clin Endocrinol Metab 2025 doi: 10.1210/clinem/dgaf498;online ahead of print
- Phase 3 trial of crinecerfont in adult congenital adrenal hyperplasia. N Engl J Med 2024 391(6):504-514
Collaborators
- Martha Quezado, MD, Laboratory of Pathology, Center for Cancer Research, NCI, Bethesda, MD
- Ninet Sinaii, PhD, MPH, Biostatistics and Clinical Epidemiology Service, NIH Clinical Center, Bethesda, MD
Contact
For more information, email dmerke@nih.gov or visit https://irp.nih.gov/pi/deborah-merke.