
Childhood Neurodegenerative Lysosomal Storage Disorders
- Anil B. Mukherjee,
MD, PhD, Head, Section on Developmental Genetics - Abhilash P. Appu, PhD, Staff Scientist
- Mannoor S. Ajayan, PhD, Visiting Fellow
- Raghavan Kutty Mahadevan, PhD, Visiting Fellow
- KC Gopal, MD, Contract Biologist
- Nisha Plavelil, PhD, Contract Biologist

We conduct both basic and translational research into a group of hereditary childhood neurodegenerative lysosomal storage disorders (LSDs) called neuronal ceroid lipofuscinoses (NCLs), commonly known as Batten disease. Loss-of-function mutations in at least 13 different genes (CLNs) underlie various types of NCL. Several years ago, we initiated investigations on CLN1 disease formerly called infantile neuronal ceroid lipofuscinosis (INCL). INCL is caused by inactivating mutations in the CLN1 gene, which encodes palmitoyl-protein thioesterase-1 (PPT1). PPT1 depalmitoylates S-palmitoylated proteins (constituents of ceroid). Numerous proteins, especially in the brain, require S-palmitoylation (also called S-acylation). It is the only reversible post-translational lipid-modification of proteins in which a saturated fatty acid (generally palmitate) is attached to specific cysteine residues in polypeptides via thioester linkage. While S-palmitoylation plays important roles in membrane anchorage of soluble proteins, protein-protein interactions, protein trafficking, and protein stability, such lipid-modified proteins must also be depalmitoylated for recycling or degradation and clearance by lysosomal hydrolases. Dynamic S-palmitoylation (palmitoylation-depalmitoylation) requires coordinated actions of two types of enzyme with opposing functions. The enzymes that catalyze S-palmitoylation are palmitoyl acyltransferases (PATs), which are zinc-finger proteins containing a common DHHC (Asp-His-His-Cys) motif. These enzymes are called ZDHHC PATs or simply ZDHHCs. The mammalian genome encodes a family of 23 ZDHHC PATS. Similarly, the PPTs are localized either in the lysosomes or in the cytoplasm. Recently, a group of cytosolic depalmitoylases, called ABHD17, were identified, which catalyze the turnover of N-Ras (a GTPase signal-transduction protein).
The aim of our translational research is to apply the knowledge gained from our basic laboratory investigations to develop novel therapeutic strategies for Batten disease. The results of our earlier investigations on CLN1 disease led to one of the first bench-to-bedside clinical trials at the NIH Clinical Center to determine whether a combination of cysteamine bitartrate (Cystagon) and N-acetylcysteine (mucomist) is beneficial for patients with this disease. Using Cln1–knockout (Cln1–/–) mice, which mimic human CLN1 disease, we discovered that PPT1 deficiency causes endoplasmic-reticulum (ER) and oxidative stresses, which, at least in part, cause neuronal death by apoptosis. During the past several years, we also delineated a mechanism by which PPT1 deficiency disrupts the recycling of synaptic vesicle (SV) proteins, which are essential for generating fresh SVs to replenish the SV pool size at the nerve terminals so as to maintain uninterrupted neurotransmission. We also discovered that the ER– and oxidative stress contribute to neuronal apoptosis and neuro-inflammation in CLN1 disease. Further, we found that PPT1 deficiency causes misrouting of the V0a1 subunit of v-ATPase (the proton pump on lysosomal membrane), dysregulating lysosomal acidification, which causes elevated pH and thus, adversely affects lysosomal degradative function.
We also developed a non-invasive method, using MRI and MRS (magnetic resonance spectroscopy) to evaluate the progression of neurodegeneration in Cln1–/– mice. The method permits repeated evaluation of potential therapeutic agents in treated animals. Application of such methods in our clinical trial with CLN1 disease also allowed us to evaluate the progressive decline in brain volume and neurodegeneration. In collaboration with other investigators, we are conducting studies to determine whether electro-retinography can be used to assess the progressive retinal deterioration in Cln1–/– as well as in Cln1 knock-in (KI) mice, which carry the nonsense mutation in the CLN1 gene commonly found in the CLN1–disease patient population in the US. Moreover, we discovered that the blood-brain barrier is disrupted in Cln1–/– mice and that this pathology is ameliorated by treatment with resveratrol, which has antioxidant properties. More recently, we discovered that a nucleophilic small molecule with antioxidant properties, N-(tert-butyl) hydroxylamine (NtBuHA), ameliorates the neurological abnormalities in Cln1–/– mice and extends their lifespan. This compound is currently undergoing preclinical evaluation for approval as an investigational new drug (IND) status by the FDA. Intriguingly, we also discovered that in Cln3–/– mice the lysosomes contain insufficient amounts of PPT1 protein and PPT1 enzymatic activity, contributing to neuropathology in this disease. These and related studies provide insight into the complex mechanisms of heritable disorders of neurodegeneration such as CLN1 disease, as well as CLN3 disease, and identify several potential therapeutic targets. Our results suggest that thioesterase-mimetic small molecules such as NtBuHA are potential therapeutics for CLN1 disease and may even be applicable to CLN3 disease. More recently, we discovered that cathepsin D (CD) deficiency in lysosomes is a common pathogenic link between CLN1 disease and CLN10 disease. Our ongoing laboratory and translational investigations are attempting to advance our knowledge of CLN1, CLN3, and CLN10 diseases.
Lysosomal cholesterol mediates mTORC1 hyperactivation contributing to neurodegeneration in CLN1 disease model.
S-palmitoylation of proteins confers hydrophobicity, increases membrane affinity, and promotes protein-protein interactions. Moreover, dynamic S-palmitoylation (palmitoylation-depalmitoylation) facilitates endosomal protein trafficking. Despite the discovery that inactivating mutations in the CLN1 gene encoding PPT1 cause CLN1 disease, a clear picture of its pathogenic mechanism has not emerged for more than two decades. In addition to its degradative function, the lysosome plays a pivotal role in cholesterol homeostasis. It is the major cellular sorting station for dietary cholesterol. Cholesterol is transported to the late endosome/lysosome and is exported to diverse cellular compartments, including to the plasma membrane and the ER. The Niemann Pick C1 (NPC1) protein, localized to the lysosomal limiting membrane, plays a critical role in sterol trafficking, and its inactivation causes the hereditary neurodegenerative lipid-storage disorder Niemann-Pick type C (NPC). Recently, it was reported that cells from patients with NPC have elevated cholesterol on the lysosomal limiting membrane, which mediates the activation of the mechanistic (mammalian) target of rapamycin complex 1 (mTORC1) protein kinase. The mTORC1 kinase integrates intracellular as well as environmental cues to regulate cell growth and metabolism. Aberrant activation of mTORC1 signaling negatively regulates autophagy, which is the principal pathway for lysosomal degradation and clearance of abnormal protein aggregates and damaged organelles. Remarkably, in all three types of autophagy, the lysosome plays pivotal roles in the degradation of cargo contained in the autophagosomes. Most notably, the dysregulation of autophagy has been implicated not only in the pathogenesis of common neurodegenerative diseases such as Alzheimer’s and Parkinson’s, but also in most of the LSDs, in which neurodegeneration is a frequent manifestation.
To understand the mechanism of CLN1 pathogenesis, we used the Cln1–/– mouse. In the brain of these mice, total cholesterol levels have been reported to be significantly higher than those in their wild-type (WT) littermates. However, in that study the cholesterol levels in lysosomes were not measured. Thus, we first sought to determine the cholesterol levels in total homogenates of cortical tissues from 2-, 4- and 6-month-old WT and Cln1–/– mice, as well as in lysosomal fractions from those tissues. We found that cholesterol levels in total lysates as well as in lysosomal fractions from cortical tissues of Cln1–/– mice in all three age groups were significantly higher than those in their WT littermates. These results were further confirmed by confocal imaging in neurons from Cln1–/– mice, which showed a substantially higher level of colocalization of Filipin III–stained cholesterol with lysosomes, which we stain with lysotracker red. Taken together, these results raised the possibility that, in the brain of Cln1–/– mice, lysosomal cholesterol homeostasis is dysregulated. Cholesterol enters the cell in its esterified form packaged with lipoproteins via the low-density lipoprotein (LDL) receptor (LDLR). Thus, dietary cholesterol enters the cell by a receptor-mediated pathway. Once within the cell, cholesterol esters are hydrolyzed by lysosomal acid lipase, liberating cholesterol, which is then transported to various cellular structures, including the ER and the plasma membrane. The NPC1 and NPC2 proteins mediate lysosomal cholesterol egress and import, respectively. Moreover, the lysosomal integral membrane protein (LIMP) 2/SCARB2 has also been reported to bind cholesterol and, like NPC1, transports cholesterol through a trans-glycocalyx tunnel, a membrane domain rich in glycoproteins and glycolipids. Under normal circumstances, the balance between the export and import maintains lysosomal cholesterol homeostasis. Thus, inactivating mutations in either the NPC1 or NPC2 gene dysregulate cellular cholesterol homeostasis, causing neurological dysfunction, leading to the fatal neurodegenerative Niemann-Pick type C disease.
We found that NPC1 protein requires dynamic S-palmitoylation (palmitoylation-depalmitoylation) for trafficking to the lysosomal membrane. Intriguingly, in Cln1–/– mice, NPC1 mistargeting to the plasma membrane caused increased oxysterol-binding protein (OSBP) on lysosomal membrane, activating cholesterol-mediated mTORC1 signaling. Activated mTORC1 signaling suppressed autophagy, contributing to neurodegeneration. Importantly, treatment of Cln1–/– mice with OSW1, a pharmacological inhibitor of OSBP, suppressed mTORC1 activation, rescued autophagy, and ameliorated neuropathology. Our findings reveal a previously unrecognized role of CLN1/PPT1 in the pathogenesis of CLN1 disease and suggest that suppression of cholesterol-mediated activation of mTORC1 signaling is a targetable pathway for CLN1 disease.
Dysregulated lysosomal Ca2+ homeostasis contributes to pathogenesis in a CLN1 disease model.
The lysosome is an organelle long known for mediating degradation and clearance of cellular waste. In recent years, it has become evident that it is a highly dynamic structure, which also plays important roles in cell metabolism in response to environmental cues. Impaired lysosomal degradative function leads to a family of about 60 inherited LSDs. Dysregulation of cellular Ca2+ homeostasis is reported to play important roles in the pathogenesis of several human diseases, including the LSDs. Defective lysosomal Ca2+ homeostasis has also been reported to impair autophagy. In most LSDs, defective autophagy leads to neurodegeneration.
The ER is the major Ca2+ repository in the cell, and Ca2+ plays a key regulatory role in autophagy, an intracellular degradative process that requires Ca2+–dependent lysosomal hydrolases for the degradation and clearance of the cargo contained in the autophagosomes. Lysosomal Ca2+ homeostasis is mediated by inositol 3-phosphate receptor 1(IP3R1)–mediated transport of Ca2+ from the ER to the lysosome. It has also been reported that selective interaction of IP3Rs with the ER–lysosome contact sites is required for the delivery of Ca2+ to the lysosome. Moreover, antagonists of IP3Rs rapidly and completely block lysosomal Ca2+ refilling. Interestingly, IP3R1 has been reported to undergo S-palmitoylation for regulating Ca2+ flux in immune cells. Furthermore, disruption of Ca2+ homeostasis may dysregulate neurotransmitter release, contributing to neurodegeneration. Autophagy is impaired by dysregulation of Ca2+ homeostasis in many LSDs, including in Cln1–/– mice. We sought to test the hypothesis that CLN1 mutations dysregulate lysosomal Ca2+ homeostasis and suppress the catalytic activities of Ca2+–dependent lysosomal hydrolases, which impair the degradation of undigested cargo in autophagosomes, causing neuropathology in CLN1 disease.
We sought to determine the mechanism by which PPT1 deficiency impairs lysosomal degradative function and contributes to CLN1 disease pathogenesis. We found that, in Cln1–/– mice, low levels of IP3R1 dysregulate lysosomal Ca2+ homeostasis. Intriguingly, the transcription factor NFATC4, which regulates IP3R1 expression, required S-palmitoylation for trafficking from the cytoplasm to the nucleus. We identified two palmitoyl acyltransferases, ZDHHC4 and ZDHHC8, which catalyzed S-palmitoylation of NFATC4. Notably, in Cln1–/– mice, reduced ZDHHC4 and ZDHHC8 levels markedly lowered S-palmitoylated NFATC4 (active) in the nucleus, which inhibited IP3R1 expression, thereby dysregulating lysosomal Ca2+ homeostasis. Consequently, Ca2+–dependent lysosomal enzyme activities were markedly suppressed. Impaired lysosomal degradative function impaired autophagy, which caused lysosomal storage of undigested cargo. Importantly, IP3R1 overexpression in Cln1–/– mouse fibroblasts ameliorated this defect. Our results reveal a previously unrecognized role of Cln1/Ppt1 in regulating lysosomal Ca2+ homeostasis, and they suggest that the defect contributes to pathogenesis of CLN1 disease.
Figure 1. Dysregulation of cholesterol homeostasis in a CLN1 disease model: misrouted Niemann-Pick C1 causes lysosomal cholesterol-mediated hyperactivation of mTORC1 kinase, contributing to neurodegeneration in CLN1 disease.
Left panel: Lysosomal cholesterol homeotasis
Schematic explaining lysosomal cholesterol homeostasis. Dietary LDL cholesterol entering the lysosome is bound to Niemann Pick 2 (NPC2) protein. Cholesterol bound to NPC2 is then transferred to NPC1 localized to the lysosomal limiting membrane. NPC1 effluxes cholesterol from the lysosomal lumen to the cytoplasm to maintain homeostasis.
Right panel: Impaired cholesterol homeostasis in CLN1 disease
WT: Niemann pick C1 (NPC1) undergoes S-palmitoylation on Cys97. Endosomal trafficking of NPC1 in WT cells is facilitated by dynamic S-palmitoylation (S-palmitoylation-depalmitoylation) on Cys97 requiring thioesterase activity of Ppt1. S-palmitoylation of NPC1 allows its binding to adaptor protein 2 (AP2). However, for its handover from AP-2 to AP3 requires depalmitoylation to detach from AP-2 and re-palmitoylation for binding to AP-3. The AP-3 bound NPC1 is then delivered to the limiting membrane of late endosome/lysosome, where it mediates lysosomal cholesterol efflux.
Cln1-/-: In Cln1-/- cells, the lack of Ppt1 activity impairs dynamic S-palmitoylation of NPC1. Consequently, NPC1 bound to AP-2 is not handed over to AP-3 and S-palmitoylated NPC1 is transported via recycling endosomes to the plasma membrane, instead of its normal location on lysosomal membrane. Misrouting of NPC1 dysregulates lysosomal cholesterol homeostasis and results in cholesterol-mediated hyperactivation of mTORC1 kinase, which suppresses autophagy, causing neurodegeneration in Cln1-/- mice.
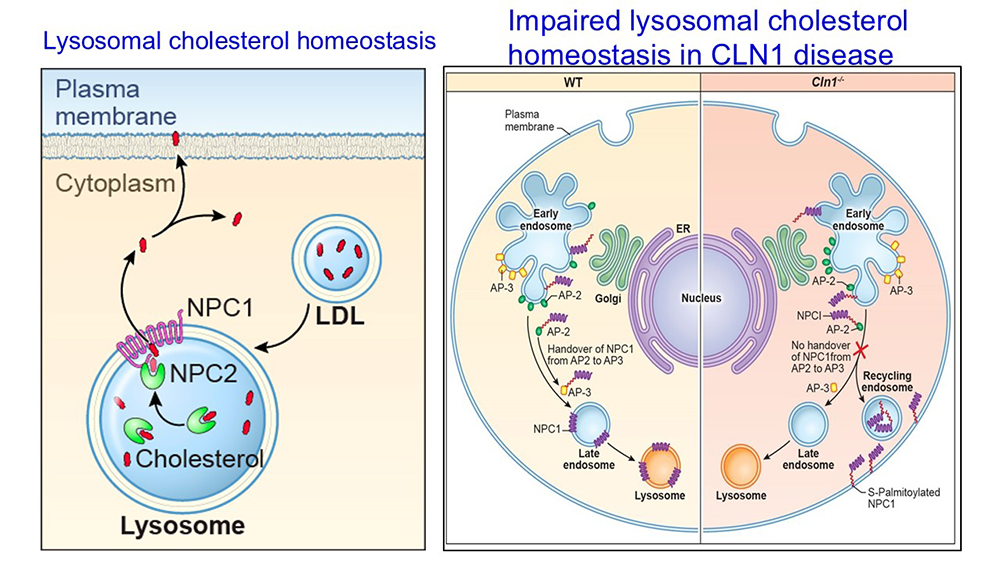
Figure 1. Dysregulation of cholesterol homeostasis in a CLN1 disease model: misrouted Niemann-Pick C1 causes lysosomal cholesterol-mediated hyperactivation of mTORC1 kinase, contributing to neurodegeneration in CLN1 disease.
Left panel: Lysosomal cholesterol homeotasis
Schematic explaining lysosomal cholesterol homeostasis. Dietary LDL cholesterol entering the lysosome is bound to Niemann Pick 2 (NPC2) protein. Cholesterol bound to NPC2 is then transferred to NPC1 localized to the lysosomal limiting membrane. NPC1 effluxes cholesterol from the lysosomal lumen to the cytoplasm to maintain homeostasis.
Right panel: Impaired cholesterol homeostasis in CLN1 disease
WT: Niemann pick C1 (NPC1) undergoes S-palmitoylation on Cys97. Endosomal trafficking of NPC1 in WT cells is facilitated by dynamic S-palmitoylation (S-palmitoylation-depalmitoylation) on Cys97 requiring thioesterase activity of Ppt1. S-palmitoylation of NPC1 allows its binding to adaptor protein 2 (AP2). However, for its handover from AP-2 to AP3 requires depalmitoylation to detach from AP-2 and re-palmitoylation for binding to AP-3. The AP-3 bound NPC1 is then delivered to the limiting membrane of late endosome/lysosome, where it mediates lysosomal cholesterol efflux.
Cln1-/-: In Cln1-/- cells, the lack of Ppt1 activity impairs dynamic S-palmitoylation of NPC1. Consequently, NPC1 bound to AP-2 is not handed over to AP-3 and S-palmitoylated NPC1 is transported via recycling endosomes to the plasma membrane, instead of its normal location on lysosomal membrane. Misrouting of NPC1 dysregulates lysosomal cholesterol homeostasis and results in cholesterol-mediated hyperactivation of mTORC1 kinase, which suppresses autophagy, causing neurodegeneration in Cln1-/- mice.
CLN1 gene mutations disrupt the nutrient-sensing scaffold on lysosomes, contributing to CLN1 disease pathogenesis.
Sensing of essential nutrients has emerged as an important function of the lysosome in coordinating cellular metabolism and growth. Signals from nutrients such as glucose, amino acids, fatty acids, and cholesterol are integrated by the lysosome, turning the cellular events from anabolic to catabolic processes such as autophagy. Whereas materials from extracellular sources are transported to the lysosome by endocytosis, those originating from intracellular sources are delivered by autophagy. Notably, activation of the mTORC1 pathway, situated at the crossroads of nutrient signaling, suppresses autophagy. The loss of autophagy in the central nervous system has been reported to cause neurodegeneration. There are three types of autophagy: macroautophagy, microautophagy, and chaperone-mediated autophagy, in all of which the lysosome plays a pivotal role in degrading the cargo contained in the autophagosomes. NCLs mostly affect children and, despite the discovery more than two decades ago that the LSD CLN1 disease is caused by inactivating mutations in the CLN1 gene, the mechanism of its pathogenesis has remained elusive. Children afflicted with CLN1 disease are phenotypically normal at birth, but by 6–18 months of age manifest psychomotor retardation. Around two years of age, they develop complete retinal degeneration, causing blindness. At around four years of age, an isoelectric electroencephalogram (EEG) attests to a vegetative state. They remain in this condition for several more years before eventual death. These grim facts underscore an urgent need for understanding the mechanism underlying pathogenesis of CLN1 disease, which may facilitate the development of an effective treatment.
Dynamic S-palmitoylation (palmitoylation-depalmitoylation) facilitates endosomal trafficking and the localization of many proteins, especially in the brain. While S-palmitoylation is catalyzed by palmitoyl acyltransferases (ZDHHC-PATs or simply ZDHHCs), depalmitoylation is mediated by thioesterases. Inactivating mutations in the CLN1 gene causing PPT1 deficiency result in lysosomal accumulation of autofluorescent ceroid lipofuscin. When the ceroid lipofuscins are organized within lysosomes, they are called GRODS (granular osmiophilic deposits), a characteristic finding in neurons and other cell types from patients with CLN1 disease. The lysosomes are the dynamic regulators of the function of many proteins, especially in the brain, and their importance is underscored by the fact that impaired lysosomal function contributes to the pathogenesis of the LSDs. These inherited diseases are characterized by metabolic dysfunction, neurodegeneration, and shortened lifespan. Moreover, it has been suggested that the lysosomal pathway plays critical roles in many cellular functions, including signaling in response to environmental cues. We reasoned that impaired dynamic S-palmitoylation of important proteins that are likely substrates of PPT1 may impair their ability to traffic to their destination. This abnormality may result in varying impairment of functions of these proteins, cumulatively contributing to neurodegeneration.
We used tissues from the cerebral cortex of Cln1–/– mice because, in our pilot study (www.clinicaltrials.gov; NCT00028262), MRI of the brain of children with CLN1 disease showed rapid degeneration of cortical tissues. We also used cultured cells from patients with CLN1 disease to determine whether the loss of CLN1/PPT1 causes aberrant activation mTORC1 signaling and suppresses autophagy. We found that, in the brain of Cln1–/– mice, Ppt1 deficiency caused aberrant activation of mTORC1, which suppressed autophagy, contributing to neurodegeneration. Emerging evidence indicates that sensing essential nutrients is an important function of the lysosome. Intriguingly, Ppt1 deficiency disrupted the lysosomal nutrient-sensing scaffold (LNSS) to which mTORC1 must attach to activate. Despite this defect, mTORC1 was activated by IGF1 (insulin-like growth factor 1) via the PI3K/Akt–mediated pathway. Importantly, treatment of Cln1–/– mice with pharmacological inhibitors of PI3K/Akt suppressed mTORC1 activation, restored autophagy and improved motor function. Our findings reveal a previously unrecognized mechanism by which Cln1/Ppt1 deficiency contributes to pathogenesis of CLN1 disease.
Dysregulated ER–Golgi protein-trafficking contributes to ER stress in a CLN1 disease model.
PPT1 catalyzes depalmitoylation of S-palmitoylated proteins for their degradation and clearance. While palmitoyl-acyltransferases (called ZDHHCs) catalyze S-palmitoylation, depalmitoylation is mediated by palmitoyl-protein thioesterases (PPTs). We previously reported that in Cln1-/- mice ER stress contributes to neurodegeneration. However, the mechanism underlying ER stress has remained elusive. Newly synthesized proteins in the ER are transported to the Golgi via COPII (coat protein complex II) vesicles, whereas the retrograde transport (Golgi to ER) is mediated by COPI vesicles. We reasoned that defective vesicular trafficking of proteins from the ER to the Golgi may lead to ER stress in Cln1-/- mice. We found that the levels of five COPII vesicle–associated proteins (i.e., Sar1, Sec23, Sec24, Sec13, and Sec31) are significantly higher in the ER fractions of cortical tissues from Cln1-/- mice compared with those from their WT littermates. Remarkably, all COPII proteins, except Sec13, undergo S-palmitoylation. Moreover, CLN8, a Batten disease-protein, also requires dynamic S-palmitoylation (palmitoylation-depalmitoylation) for ER–Golgi trafficking. Intriguingly, Ppt1 deficiency in Cln1-/- mice impairs ER–Golgi trafficking of Cln8 protein along with several COPII–associated proteins. We propose that defective vesicular trafficking from the ER to Golgi causes excessive accumulation of proteins in the ER, contributing to ER stress in CLN1 disease.
Mediators of neuroinflammation in a CLN1 disease model resemble those in common neurodegenerative disorders.
Neuroinflammation contributes to neurodegeneration, a devastating manifestation in most of the 70 LSDs. NCLs represent a group of the most common neurodegenerative LSDs. As stated in the introduction, inactivating mutations in 13 different genes (called CLNs) underlie various types of NCLs. The CLN1 disease is caused by inactivating mutations in the CLN1 gene encoding palmitoyl-protein thioesterase-1 (PPT1). Despite this knowledge, the mechanism underlying neuroinflammation in CLN1 disease has remained elusive. Genetic studies in a growing number of neurodegenerative diseases have linked activated microglia with neuroinflammation. Intriguingly, an interplay between the disease-associated microglia apolipoprotein E (ApoE) and triggering receptor expressed on myeloid cells 2 (Trem2) is reported to mediate neuroinflammation in Alzheimer’s disease (AD). We tested a hypothesis that neuroinflammatory mediators in a mouse model of CLN1 disease bear similarities to those in common neurodegenerative disorders such as Alzheimer’s and Parkinson’s. To test our hypothesis, we used Cln1–/– mice, a reliable animal model of human CLN1 disease. In most of the experiments, we used brain tissues from 2-, 4- and 6-month-old Cln1–/– mice and their wild-type (WT) littermates. We used molecular, biochemical, immunohistochemical, and cell-biological methods to evaluate the levels of inflammatory mediators in these mice. We found that, in the brain of Cln1–/–, mice the mRNA and protein levels of ApoE and Trem2 are overexpressed. Intriguingly, the transcription factors PU.1 and Irf8, which regulate the generation of microglia, are also overexpressed in the Cln1–/– mouse brain. Remarkably, the levels of neuroinflammatory mediators such as GFAP, lipocalin2, Glut1, aquaporin-4, Glt1, p-tau, ELOVL1, cGAS/STING, ceramide, and complement C3 are markedly elevated in the Cln1–/– mouse brain. Previously, we reported that ER– and oxidative stress contributes to neuronal death in Cln1–/– mice. Consistent with the finding of high oxidative stress, the astrocytes in Cln1–/– mice contain high levels of lipid droplets. Our findings reveal a striking similarity between the levels of various of neuroinflammatory mediators in a monogenic neurodegenerative LSD and those reported in common neurodegenerative disorders in which underlying genetic links are often difficult to ascertain.
Additional Funding
- Dr. Abhilash P. Appu, a Staff Scientist has received an Early Career Award of $25,000.00
Publications
- Disruption of lysosomal nutrient sensing scaffold contributes to pathogenesis of a fatal neurodegenerative lysosomal storage disease [Editor's Choice]. J Biol Chem 2024 300(2):105641
- Emerging new roles of the lysosome and neuronal ceroid lipofuscinoses. Mol Neurodegener 2019 14(1):4
- In a mouse model of INCL reduced S-palmitoylation of cytosolic thioesterase APT1 contributes to microglia proliferation and neuroinflammation. J Inherit Metab Dis 2021 44:1051–1069
- Cln1-mutations suppress Rab7-RILP interaction and impair autophagy contributing to neuropathology in a mouse model of INCL. J Inherit Metab Dis 2020 43:1082–1101
- Niemann Pick C1-mistargeting disrupts lysosomal cholesterol homeostasis contributing to neurodegeneration in a Batten disease model. Sci Adv 2024 under revision
Collaborators
- Aiyi Liu, PhD, Biostatistics & Bioinformatics Branch, NICHD, Bethesda, MD
- Rafael M. Previde, PhD, Section on Cellular Signaling, NICHD, Bethesda, MD
- Stanko Stojilkovic, PhD, Section on Cellular Signaling, NICHD, Bethesda, MD
- Wadih Zein, MD, Ophthalmic Genetics and Visual Function Branch, NEI, Bethesda, MD
Contact
For more information, email mukherja@exchange.nih.gov or visit https://www.nichd.nih.gov/research/atNICHD/Investigators/mukherjee.