
Gene Regulation in Innate Immunity

- Keiko Ozato,
PhD, Head, Section on Molecular Genetics of Immunity - Anup Dey, PhD, Biologist
- Tiyun Wu, PhD, Staff Scientist
- Sakshi Chauhan, PhD, Visiting Fellow
- Kentaro Hoshi, MD, PhD, Visiting Fellow
- Fuki Kudoh, PhD, Visiting Fellow
- Eunju Lee, PhD, Visiting Fellow
- Keita Saeki, MD, PhD, Visiting Fellow
- Vishal Nehru, PhD, Contractor
The laboratory is interested in the role of chromatin and gene regulation in innate immunity. We study three nuclear factors: histone H3.3, BRD4, and IRF8. Histone H3.3 is a variant histone that is incorporated into nucleosomes during transcriptional elongation, a defining feature of this variant. Other canonical core histones are deposited into nucleosomes during replication. For this reason, H3.3 is thought to be involved in epigenetic memory created after transcription, although experimental evidence for memory formation/maintenance is scant. BRD4 is a bromodomain protein of the BET (bromodomain and extra-terminal domain) family, expressed broadly in many cells, from early embryos to adults. Through the bromodomain, BRD4 binds to acetylated but not to unacetylated histones. BRD4 is thus called a “chromatin reader,” a type of regulatory factor capable of conveying epigenome information to gene expression. Furthermore, BRD4 binds to the elongation factor complex P-TEFb through the C-terminal domain, and drives transcription of many genes by making RNA polymerase II move through the gene body, generating nascent mRNA. Many recent reports point out that BRD4 promotes growth of cancer cells, including various blood cancers, by mediating the formation of super enhancers involved in cell-cycle progression. IRF8, which we discovered in 1990, is a DNA–binding transcription factor that plays an essential role in innate resistance against a wide array of pathogens (Figure 1A for its structure). IRF8 is expressed mostly in cells of the myeloid lineage, including monocytes/macrophages, dendritic cells, and microglia. IRF8 is strongly induced when stimulated by interferons (IFN). In addition, it is upregulated when myeloid cells encounter pathogen-derived molecules and agents produced by stress. In turn, IRF8 activates many genes important for host resistance. IRF8–induced genes include those involved in autophagy and lysosome-mediated pathogen clearance; IRF8 does so by binding to small DNA motifs present in promoter and enhancer regions of the target genes.
Figure 1. Structure of IRF8
IRF8 is a transcription factor of the IRF family. It has the DNA–binding domain in the N-terminus and an the IAD–regulatory domain in the C-terminus; through the latter it interacts with other factors.
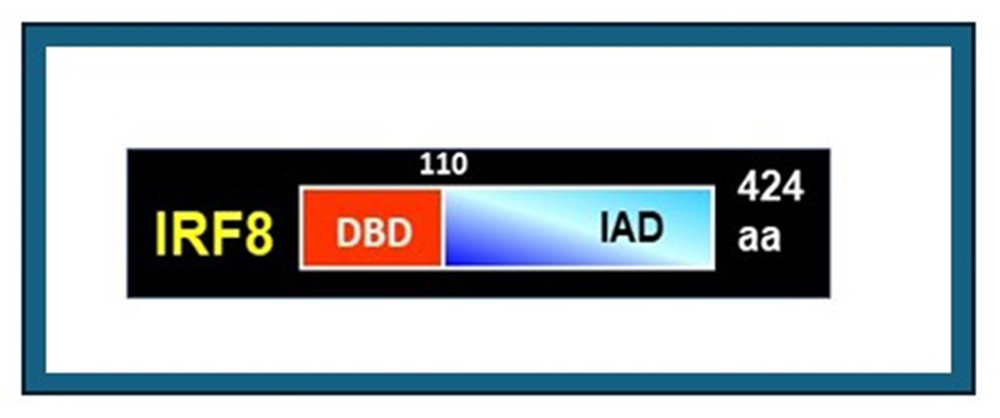
Figure 1. Structure of IRF8
IRF8 is a transcription factor of the IRF family. It has the DNA–binding domain in the N-terminus and an the IAD–regulatory domain in the C-terminus; through the latter it interacts with other factors.
Gene regulation in the innate immunity
Our laboratory has been interested in chromatin and gene regulation in innate immunity for more than forty years. Innate immunity represents natural resistance to pathogens. Innate immunity is older than adaptive immunity in evolution, but its mechanisms are not fully understood. To gain insight into how innate immunity works, we study the role of three nuclear factors: IRF8, BRD4, and histone H3.3. IRF8 is a DNA–binding transcription factor which we discovered in 1990 (Figure 1). IRF8 is expressed mostly in cells of the myeloid lineage, critical for innate immunity, including monocytes/macrophages, dendritic cells, and microglia. IRF8 directs the development of these cells from myeloid progenitors. IRF8 binds to the DNA motif termed ISRE. IRF8 heterodimerizes with PU.1, an Ets–family (erythroblast transformation–specific) transcription factor, and binds to ETS/IRF composite elements. These elements are present in the enhancer regions of many genes critical for monocyte and dendritic cell development. IRF8 also binds to interferon-stimulated genes (ISGs), which provide resistance to pathogens. We constructed Irf8 constitutive knockout (KO), Irf8 conditional KO (cKO), and GFP (green fluorescent protein) reporter mice. Studies with these mice confirmed that IRF8 plays an essential role in innate resistance against a whole array of pathogens. Analysis of IRF8KO and IRF8cKO mice also revealed that IRF8 regulates proliferation of myeloid progenitor cells and acts as a leukemia suppressor. Furthermore, genome-wide association studies (GWAS) found IRF8 to be a risk factor for diseases such as leukemia, autoimmune diseases (systemic lupus erythematosus [SLE]), multiple sclerosis (MS), rheumatoid arthritis (RA), and cardiovascular diseases. Overall, GWAS data point to a central role of IRF8 in promoting host resistance and inflammation.
As stated above, BRD4 is a bromodomain protein of the BET family that binds to acetylated histones (Figure 2). In addition, it recruits the elongation factor P-TEFb via the C-terminal domain and promotes transcription of many cellular genes carrying acetylated histones. BRD4 is expressed at high levels in most, if not all cells, and is essential for early embryogenesis and postnatal development. Compelling evidence indicates that BRD4 promotes cancer growth. Small-molecule inhibitors (BETi). which block binding of BRD4 to acetylated histones, arrest cancer growth. More recently, another class of BRD4 inhibitors that degrade BRD4 have been developed, which likewise inhibit cancer growth. These findings define BRD4 as a cancer promoter. However, whether and how BRD4 regulates growth of normal cells in vivo has remained elusive.
Figure 2. Structure of BRD4
BRD4 is a 200 Kda nuclear protein with two distinct domains, a bromodomain through which it interacts with acetylated histones, and the ET domain through which it binds to elongation factor PTEFb to directs transcription elongation of many genes.

Figure 2. Structure of BRD4
BRD4 is a 200 Kda nuclear protein with two distinct domains, a bromodomain through which it interacts with acetylated histones, and the ET domain through which it binds to elongation factor PTEFb to directs transcription elongation of many genes.
As stated above, Histone H3.3 is a conserved variant histone that is incorporated into nucleosomes during transcriptional elongation, a defining feature of this variant. Structurally, H3.3 differs from core histone H3.1 and H3.2 in only a few amino acids. H3.3 localizes mostly to the euchromatin, as opposed to H3.1 and H3.2, which also localize to heterochromatin (Figure 3). Despite its presumed importance, the role of H3.3 in the development of immune cells, both innate and adaptive, has remained unknown.
Figure 3. Nuclear localization of the variant histone H3.3
Top: Canonical histone H3 (H3.1 and H3.2), synthesized and incorporated into nucleosomes during replication. It localizes to both euchromatin and heterochromatin.
Bottom: Variant histone H3.3 synthesized throughout the cell cycle is incorporated into nucleosomes during transcription. H3.3 localizes strongly to euchromatin.
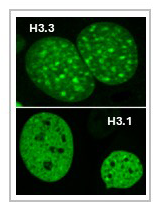
Figure 3. Nuclear localization of the variant histone H3.3
Top: Canonical histone H3 (H3.1 and H3.2), synthesized and incorporated into nucleosomes during replication. It localizes to both euchromatin and heterochromatin.
Bottom: Variant histone H3.3 synthesized throughout the cell cycle is incorporated into nucleosomes during transcription. H3.3 localizes strongly to euchromatin.
IRF8 sets epigenome structure for microglia and confers their function.
As previously noted, IRF8 is highly expressed in microglia, which led us investigate the role of IRF8 in these cells. Microglia are the sole cell type in the brain that protect from pathogen infection and restore neuronal injury. Microglia, unlike neurons, originate from the progenitor in the yolk sac, and then move into the brain. Microglia regulate the development of neuronal synapses. Recent studies demonstrated that microglia are a main driver of Alzheimer’s disease (AD), although they control the disease at an early stage. We found that Irf8 KO microglia have abnormal morphology but do not express a number of microglia-specific surface markers and other genes that define microglia (Figure 4). Thus, Irf8KO and irf8cKO cells do not survey the brain and fail to provide innate resistance to incoming pathogens. Furthermore, we found that AD progresses more slowly in Irf8KO. In accordance, amyloid-beta accumulates less extensively in mice without an irf8 gene. These results were reported in Reference 1.
Figure 4. IRF8 governs microglia identity.
Top: Normal microglia have ramified extensions and are highly mobile. IRF8KO microglia show activated morphology and fail to move around to find obstacles and injury.
Bottom: Single-cell (sc) RNA-Seq reveals that distinct sets of genes are expressed, constituting several clusters in both WT and IRF8KO microglia.
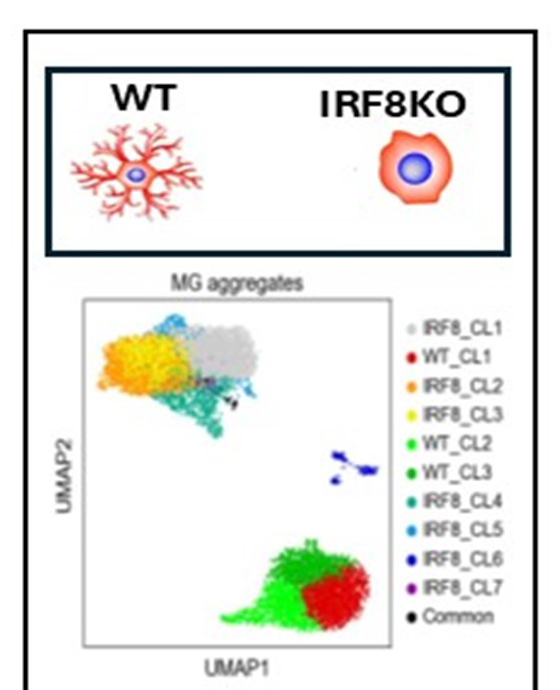
Figure 4. IRF8 governs microglia identity.
Top: Normal microglia have ramified extensions and are highly mobile. IRF8KO microglia show activated morphology and fail to move around to find obstacles and injury.
Bottom: Single-cell (sc) RNA-Seq reveals that distinct sets of genes are expressed, constituting several clusters in both WT and IRF8KO microglia.
More recently, we investigated whether IRF8 plays a role in regulating behavior. A series of behavioral tests found that Irf8KO mice exhibit an anxiety-like disorder in a sex-dependent manner. Many of them also develop obsessive compulsive behavior, a behavioral abnormality that is likely attributable to microglia lacking IRF8, given that anxiety is caused by defective neuronal connection in the brain. Besides, we found that female Irf8KO microglia produce more reactive oxygen species than do male microglia. It is possible that Irf8KO microglia have increased oxidative stress and are unable to form the proper neuronal connections necessary for controlling anxiety like behavior. This work has been submitted for publication.
BRD4 directs cell cycle progression by inhibiting endogenous DNA damage.
As noted in the Introduction, BRD4 promotes growth of many cancers. Some pharmacological inhibitors targeting BRD4 can potently arrest cancer growth. Such inhibitors increase R-loop formation and DNA damage, leading to cancer-cell growth arrest. However, it has been unclear whether BRD4 controls R-loop and DNA damage in normal cells. To gain mechanistic insight into the role of BRD4 in normal cell growth, we created mouse embryonic fibroblasts in which Brd4 was conditionally knocked out. Brd4cKO cells grew much more slowly than wild-type (WT) cells, as many cells remained at mid-to late S phase, without progressing to G2 phase; the few cells that reached G2 mostly failed to execute mitosis. Nearly 500 cell-cycle genes were downregulated in Brd4 cKO cells, including core histone genes and genes necessary for cell division. Brd4cKO cells exhibited increased R-loop formation and extensive DNA damage, seen as nuclear S9.6 signals and extensive γH2AX foci. Furthermore, genes critical for R-loop suppression were downregulated in Brd4cKO cells, including RnaseH1/2 and topoisomerases Top1/2. Likewise, genes critical for DNA damage response (DDR), including ATM, ATR, p53, p21, and p53BP1, were greatly reduced in Brd4cKO cells. ChIP-Seq analysis found that BRD4 occupied numerous cell-cycle genes as well as genes controlling R-loop formation and DDR throughout the entire cell cycle stages. The occupancy was detected on the promoter, transcription start site, and gene body. Taken together, our work demonstrates that BRD4 epigenetically marks genes controlling R-loop formation and DDR along with many cell-cycle genes and ensures ordered cell cycle events and the maintenance of genome integrity.
Live cell analysis of the histone variant H3.3
Incorporation of the variant histone H3.3 into the chromatin occurs during transcription, a defining feature of the variant. However, molecular mechanisms affecting H3.3 dynamics remain unclear. Furthermore, little is known about how H3.3 is turned over and replaced by new H3.3. We conducted fluorescence recovery after photobleaching (FRAP) to examine H3.3 mobility in mouse embryonic fibroblasts. The SNAP tag (a self-labeling protein tag) system enabled us to study the mobility of both preexisting and newly synthesized H3.3 pools. Our results showed that H3.3 is significantly more mobile than the core histone H3.1. We found that H3.3 mobility has three distinct components, fast and slow mobile components, and an immobile component, while H3.1 had a single immobile fraction. The histone H3.3 chaperons HIRA and NSD2 were necessary for H3.3 to be mobile. Strikingly, inhibition of transcriptional elongation by Flavopiridol (a cyclin-dependent kinase inhibitor) made H3.3 completely immobile, indicating that H3.3 mobility is a function of active transcription. To investigate decay kinetics of H3.3, we devised a live-cell imaging approach, in which we traced time-dependent loss of pulse-labeled SNAP signals of the H3.3. We found that SNAP–H3.3 decays with two distinct kinetics, first relatively fast decay followed by slower decay over 50 hours. The deletion of Hira and Nsd2 led to a slower decay. Surprisingly, transcription inhibition by Flavopiridol almost completely halted H3.3 decay for approximately 10–15 hours. Our results unraveled a strong link between H3.3 decay and transcriptional activity. Together our work illustrates that mobility and turn-over of H3.3 is dictated by ongoing transcription [Reference 3].
Histone H3.3 ensures cell proliferation and genomic stability during myeloid cell development.
Variant histone H3.3 is believed to be critical for survival of many cells, given that it is deposited in expressed genes through transcription, a feature different from core histones. For example, H3.3 deletion leads to early embryonic lethality in mice. However, a requirement for H3.3 in later stages of development has remained unclear. The aim of this work was to elucidate the role of H3.3 for the development of the myeloid lineage, important for innate immunity. We conditionally knocked out (cKO) the H3.3 genes in myeloid progenitor cells differentiating into bone marrow–derived macrophages (BMDMs). Progenitor cells lacking H3.3 were defective in replication, suffered from extensive DNA damage, as verified by the formation of gH2AX foci. DNA damage–response (DDR) pathways ensued in H3.3 cKO cells, which led to apoptosis of many cells. Some H3.3cKO cells survived and differentiated into BMDMs. However, these cells were not normal, as they expressed many interferon-stimulated genes (ISGs) throughout differentiation, which set the cells and their milieu in an inflammatory state (Figure 5). To clarify the mechanism by which H3.3 cKO cells activate ISGs, we tested H3.3 cKO cells lacking STING (stimulator of interferon genes) or RIG-I (retinoic acid-inducible gene I, a key sensor of virus infection) or IRF7. We found that these pathways were not responsible for the ISG induction in H3.3cKO BMDM, suggesting an alternative, previously unstudied mechanism. Overall, H3.3cKO BMDMs retained general nucleosomal structure genome-wide. In summary, H3.3 is indispensable for the development of healthy, functional macrophages. This work has been submitted to a scientific journal, and the paper is currently under revision.
Figure 5. Histone H3.3 ensures cell proliferation and genomic stability during myeloid cell development.
Top: In the 7–day in vitro culture model, bone marrow progenitor cells from wild-type (WT) proliferate in the early stage (day 3), then differentiate to become functional macrophages by day 7.
Bottom: Bone marrow progenitor cells from H3.3cKO mice succumb to DNA damage. H3.3cKO macrophages express many interferon-stimulated genes (ISGs), including inflammatory cytokines, rendering H3.3 cKO macrophages and the surrounding environment inflammatory.
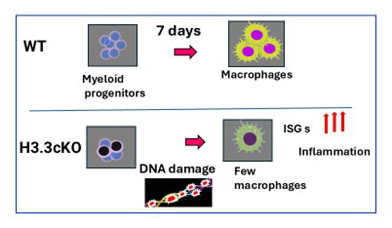
Figure 5. Histone H3.3 ensures cell proliferation and genomic stability during myeloid cell development.
Top: In the 7–day in vitro culture model, bone marrow progenitor cells from wild-type (WT) proliferate in the early stage (day 3), then differentiate to become functional macrophages by day 7.
Bottom: Bone marrow progenitor cells from H3.3cKO mice succumb to DNA damage. H3.3cKO macrophages express many interferon-stimulated genes (ISGs), including inflammatory cytokines, rendering H3.3 cKO macrophages and the surrounding environment inflammatory.
Publications
- IRF8 defines the epigenetic landscape in postnatal microglia, thereby directing their transcriptome programs. Nat Immunol 2024 10:1928-1942
- Brd4 expression in microglia exacerbates demyelination and neuroinflammation in experimental autoimmune encephalomyelitis. J Neuroinflammation 2025 22(1):148
- Nehru V, Ball D, Mukherjee A, Kurotaki D, Karpova TS, Ozato K. J Biol Chem 2025 302(6):108557
Collaborators
- Robert J. Crouch, PhD, Section on Formation of RNA, NICHD, Bethesda, MD
- Daisuke Kurotaki, PhD, Kumamoto University, Kumamoto City, Japan
- Justin Milner, PhD, University of North Carolina, Chapel Hill, NC
- Tomohiko Tamura, MD, PhD, Tokyo University, Tokyo, and Yokohama City University, Yokohama, Japan
- Vivek Thumbigere Math, DDS, PhD, University of Maryland, School of Dentistry, Baltimore, MD
Contact
For more information, email ozatok@mail.nih.gov or visit https://www.nichd.nih.gov/research/atNICHD/Investigators/ozato.