
Diagnosis, Localization, Pathophysiology, and Molecular Biology of Pheochromocytoma and Paraganglioma
- Karel Pacak,
MD, PhD, DSc, Head, Section on Medical Neuroendocrinology - Sara Talvacchio, BSN, Research Nurse
- Alberta Derkyi, CRNP, Nurse Practitioner
- Tamara Prodanov, MD, Clinical Trial Database (CTDB) Coordinator
- Thanh-Truc Huynh, MS, Biologist
- Katerina Hadrava Vanova, PhD, Postdoctoral Visiting Fellow
- Matthew Arman Nazari, MD, Postdoctoral Fellow
- Ondrej Uher, PhD, Postdoctoral Visiting Fellow
- Kailah Charles, BS, Predoctoral Fellow
- Jared Rosenblum, MD, Volunteer

Pheochromocytomas (PHEOs) and paragangliomas (PGLs) are rare but clinically important chromaffin-cell tumors that typically arise from the adrenal gland and from extra-adrenal paraganglia, respectively. The clinical features and consequences of PHEO/PGL, collectively known as PPGLs, result from overproduction and release of catecholamines (norepinephrine and epinephrine). An undetected PHEO/PGL poses a hazard to patients undergoing surgery, childbirth, or general anesthesia because of the potential for excess catecholamine secretion, which can result in significant, often catastrophic outcomes. Diagnosing and localizing a PHEO/PGL can be challenging. Plasma and urinary catecholamines, as well as their metabolites, and radio-iodinated metaiodobenzylguanidine (MIBG) scanning can yield false-positive or false-negative results in patients harboring the tumor, and computed tomography (CT) and magnetic resonance imaging (MRI) lack sufficient specificity. The molecular mechanisms by which genotypic changes predispose to the development of PHEO/PGL (PPGLs) remain unknown, even in patients with identified mutations. Moreover, in patients with hereditary predispositions, PPGLs differ in terms of their growth, malignant potential, catecholamine phenotype, responses to standard screening tests, various imaging modalities, and therefore to different therapeutic options. We focus on developmental, molecular, genetic, epigenetic, proteomic, metabolomic, immunologic, and other types of studies to investigate the bases for a predisposition to develop PPGLs and the expression of various neuro-chemical phenotypes and malignant potentials, including therapeutic responses and appropriate follow-up.
Figure 1. Immune signature of pheochromocytoma and paraganglioma (PPGL) in the context of neuroendocrine neoplasms associated with prognosis
Clustering of immune cell infiltrations on TCGA (The Cancer Genome Atlas) PPGL samples:
A. Unsupervised clustering of PPGL according to immune cell infiltrations identifies 5 clusters. Cluster 1 (red) is enriched for macrophages and cell-cycle functions; cluster 2 (gray) is enriched for monocytes; cluster 3 (green) is enriched for chemokines associated with activated natural killer (NK) cells and mast cells; cluster 4 (cyan) is associated with regulatory T cells, TGF-beta, and macrophages; cluster 5 (blue) is associated with cytotoxic CD8+ T cells.
B. Mutual co-occurrence map for PPGL immune clusters and molecular subtypes. The mutual co-occurrence p values are calculated using Fisher’s test for samples belonging to each immune cluster and molecular subtype of PPGL. PPGL patients belonging to SDHx–mutated subtype also belong to Immune cluster 1 (significant co-occurrence p < 0.05), and PPGL patients belonging to cortical admixture subtype also belong to Immune cluster 3 (significant co-occurrence p < 0.05).
C. The relative proportion of five immune clusters in known molecular subtypes of PPGL is shown by a stacked bar plot.
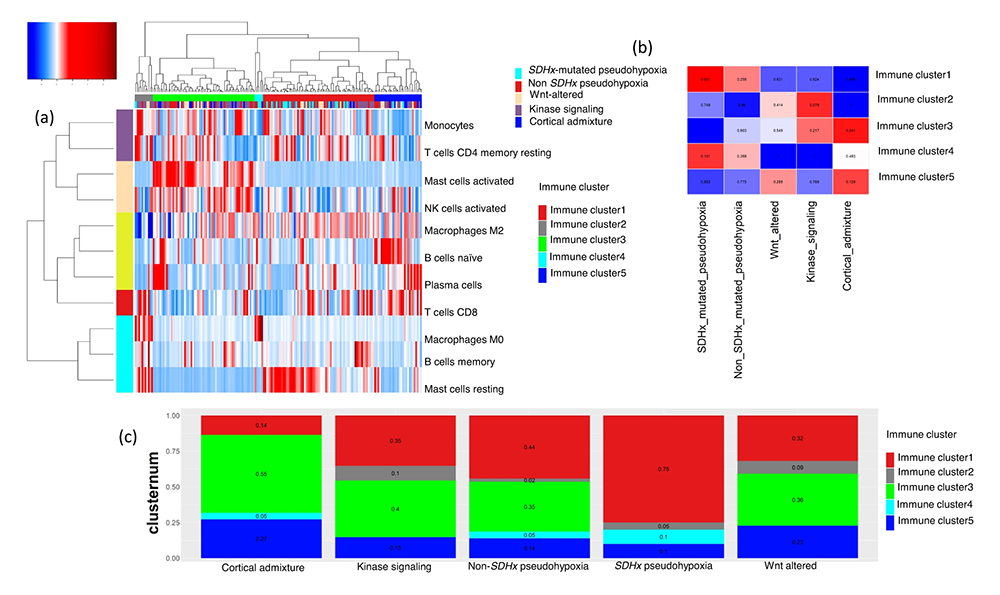
Figure 1. Immune signature of pheochromocytoma and paraganglioma (PPGL) in the context of neuroendocrine neoplasms associated with prognosis
Clustering of immune cell infiltrations on TCGA (The Cancer Genome Atlas) PPGL samples:
A. Unsupervised clustering of PPGL according to immune cell infiltrations identifies 5 clusters. Cluster 1 (red) is enriched for macrophages and cell-cycle functions; cluster 2 (gray) is enriched for monocytes; cluster 3 (green) is enriched for chemokines associated with activated natural killer (NK) cells and mast cells; cluster 4 (cyan) is associated with regulatory T cells, TGF-beta, and macrophages; cluster 5 (blue) is associated with cytotoxic CD8+ T cells.
B. Mutual co-occurrence map for PPGL immune clusters and molecular subtypes. The mutual co-occurrence p values are calculated using Fisher’s test for samples belonging to each immune cluster and molecular subtype of PPGL. PPGL patients belonging to SDHx–mutated subtype also belong to Immune cluster 1 (significant co-occurrence p < 0.05), and PPGL patients belonging to cortical admixture subtype also belong to Immune cluster 3 (significant co-occurrence p < 0.05).
C. The relative proportion of five immune clusters in known molecular subtypes of PPGL is shown by a stacked bar plot.
Clinical and genetic aspects of pheochromocytoma and paraganglioma
Patients with germline SDHD pathogenic variants (encoding succinate dehydrogenase subunit D, i.e., paraganglioma 1 syndrome) are predominantly affected by head and neck PGLs, which, in almost 20% of patients, might coexist with PGLs arising from other locations (e.g., adrenal medulla, para-aortic, cardiac or thoracic, and pelvic). Given the higher risk of tumor multifocality and bilaterality for PPGL, more because of SDHD pathogenic variants than for their sporadic and other genotypic counterparts, the management of patients with SDHD PPGLs is clinically complex in terms of imaging, treatment, and management options. Furthermore, locally aggressive disease can be discovered either at a young age or late in the disease course, which presents challenges in balancing surgical intervention with various medical and radio-therapeutic approaches. The axiom ‘first, do no harm’ should always be considered, and an initial period of observation (i.e., watchful waiting) is often appropriate to characterize tumor behavior in patients with these pathogenic variants. Such patients should be referred to specialized high-volume medical centers. Thus, we published the first international consensus guideline on the management of PPGLs in these patients [Reference 1] to help physicians with the clinical decision-making process when caring for patients with these tumors.
PPGLs have up to a 20% rate of metastatic disease, which cannot be reliably predicted. Our study prospectively assessed whether the dopamine metabolite methoxytyramine might predict metastatic disease, whether predictions might be improved using machine-learning models that incorporate other features, and how machine learning–based predictions compare with predictions made by specialists in the field. In this machine-learning modelling study, we used cross-sectional cohort data from the international trial, based in Germany, Poland, and the Netherlands, to prospectively examine the utility of methoxytyramine to predict metastatic disease in 267 patients with PPGLs and positive biochemical test results at initial screening. Another retrospective dataset of 493 patients with these tumors, enrolled under clinical protocols at National Institutes of Health (00-CH-0093) and the Netherlands (PRESCRIPT trial), was used to train and validate machine-learning models according to selections of additional features. The best performing machine-learning models were then externally validated using data for all patients in the international trial. For comparison, 12 specialists provided predictions of metastatic disease using data from the training and external validation datasets. Prospective predictions indicated that plasma methoxytyramine could identify metastatic disease at sensitivities of 52% and specificities of 85%. The best performing machine-learning model was based on an ensemble tree classifier algorithm that used nine features: plasma methoxytyramine, metanephrine, normetanephrine, age, sex, previous history of PPGL, location and size of primary tumors, and presence of multifocal disease. The model had an area under the receiver operating characteristic curve of 0·942 (95%) that was significantly larger than that of the best performing specialist (0.815) before and after (0.812) provision of SDHB variant data. Sensitivity for prediction of metastatic disease in the external validation cohort reached 83% at a specificity of 92%. We concluded that, although methoxytyramine has some utility for prediction of metastatic PPGLs, its sensitivity is limited. Predictive value is considerably enhanced with machine-learning models that incorporate our nine recommended features. Our final model provides a preoperative approach to predict metastases in patients with PPGLs, and thereby guide individualized patient management and follow-up.
Recent advances in oncology-related immunotherapy, specifically in targeting of programmed cell death-1 (PD-1)/programmed death-ligand 1 (PD-L1) pathways, have identified a new treatment potential in a variety of tumors, including advanced and rare tumors. Only a fraction of patients being treated by immune-checkpoint inhibitors have been shown to benefit from them, underscoring a need for strategies that identify patients who are most likely to show a favorable response. Building on recent, promising outcomes in a clinical study of metastatic PPGL using pembrolizumab, a humanized IgG4κ monoclonal antibody targeting the PD-1/PD-L1 pathway, we examined PD-L1 and PD-L2 expression in relation to oncogenic drivers in our PPGL patient cohort to explore whether expression can predict metastatic potential and/or be considered a predictive marker for targeted therapy. We evaluated RNA expression in the NIH cohort of 48 patients with known genetic predisposition (sporadic; pseudohypoxia: SDHB, VHL, EPAS1, EGLN1; kinase signaling: RET, NF1) and six normal medulla samples (NAM). For comparison, 72 PPGL samples from The Cancer Genome Atlas (TCGA) were used for analysis of gene expression based on the variant status (pseudohypoxia: SDHB, VHL, EPAS1, EGLN1; kinase signaling: NF1, RET). Expression of PD-L1 was higher in the PPGL cohort than in normal adrenal medulla, aligning with the TCGA analysis, whereas PD-L2 was not elevated. However, expression of PD-L1 was lower in the pseudohypoxia cluster than in the sporadic and the kinase-signaling subtype cluster, suggesting that sporadic and kinase-signaling cluster PPGLs could benefit from PD-1/PD-L1 therapy more than the pseudohypoxia cluster. Within the pseudohypoxia cluster, expression of PD-L1 was significantly lower in both SDHB– and non-SDHB–mutated tumors than in sporadic tumors. PD-L1 and PD-L2 expression were not affected by the metastatic status. We conclude that PD-L1 and PD-L2 expression in our cohort of PPGL tumors was not linked to metastatic behavior; however, the presence of PPGL driver mutation could be a predictive marker for PD-L1–targeted therapy and an important feature for further clinical studies in patients with PPGL.
Imaging of pheochromocytomas and paragangliomas
Adrenal neoplasms rarely occur in children. They can be diagnosed in the presence of endocrine, metabolic, or neurological problems, an abdominal mass, more rarely an adrenal incidentaloma (adrenal masses detected on imaging performed for reasons other than suspected adrenal disease), or in the context of an adrenal mass discovered in the evaluation of childhood cancer, including hematologic malignancy. According to standard medical practice, pediatric malignancies are almost always evaluated by 18F-fluorodeoxyglucose positron emission tomography with computed tomography ([18F]FDG PET/CT). Nuclear imaging using specific radiotracers is also an important tool for diagnosing and staging neuroblastoma, pheochromocytoma, hormone hypersecretion, or indeterminate adrenal masses. The Hippocratic oath “primum non nocere” encourages limitation of radiation in children per the ALARA concept (as low as reasonably achievable), but should not lead to the under-use of nuclear imaging because of the potential risk of inaccurate diagnosis or underestimation of the extent of disease. As in adults, nuclear imaging in children should be performed in conjunction with hormone evaluation and morphological imaging. In this imaging study, we summarized current knowledge focusing on imaging of adrenal neoplasms (including pheochromocytoma) in children.
In another study, we examined coronary arterial luminal diameter, vessel wall thickness (VWT), and plaque burden in PPGL patients and an age-matched population of healthy volunteers (HVs). Sixteen HVs and 16 PPGL patients participated in this NIH IRB–approved, HIPAA (Health Insurance Portability and Accountability Act)–compliant, prospective study. In patients with PPGLs, there was an approximate 55% reduction in luminal area without increased VWT. We concluded that PPGL patients have significantly reduced coronary luminal area, a likely consequence of catecholamine excess. The reduction in coronary luminal area, however, remained significant despite treatment with α-adrenoceptor blocking agents. Such findings suggest that the reduction in coronary arterial luminal area may be the result of excessive vasoconstriction as opposed to atherosclerosis or catecholamine-mediated arterial remodeling. Further investigation is needed, given that directed treatments such as with coronary artery vasodilators (such as with verapamil, already used to treat PPGL patients) may provide benefit.
Proteomic, metabolic, and immune aspects of pheochromocytoma and paraganglioma
New drug targets and proteins that would assist sensitive PPGL imagining could improve therapy and the quality of life of patients with PPGL, namely those with recurrent or metastatic disease. Using a combined proteomic strategy, we looked for such clinically relevant targets among integral membrane proteins (IMPs) upregulated on the surface of tumor cells and non-membrane druggable enzymes in PPGL. We conducted a detailed proteomic analysis of 22 well characterized human PPGL samples and normal chromaffin tissue from adrenal medulla. A standard quantitative proteomic analysis of tumor lysate, which provides information largely on non-membrane proteins, was accompanied by specific membrane proteome–aimed methods, namely glycopeptide enrichment using lectin-affinity, glycopeptide capture by hydrazide chemistry, and enrichment of membrane-embedded hydrophobic transmembrane segments. The study identified 67 cell-surface integral membrane proteins strongly upregulated in PPGL compared with control chromaffin tissue. We prioritized the proteins based on their already documented direct role in cancer-cell growth or progression. Increased expression of the seven most promising drug targets (CD146, CD171, ANO1, CD39, ATP8A1, ACE, and SLC7A1) were confirmed using specific antibodies. Our experimental strategy also provided expression data for soluble proteins. Among the druggable non-membrane enzymes upregulated in PPGL, we identified three potential drug targets (SHMT2, ARG2, and autotaxin) and verified their upregulated expression. We concluded that the application of a combined proteomic strategy, recently presented as “Pitchfork,” enabled quantitative analysis of both membrane and non-membrane proteome, and resulted in identification of these 10 potential drug targets in human PPGL. Seven membrane proteins localized on the cell surface and three non-membrane druggable enzyme proteins were identified and verified as significantly upregulated in PPGL. All the proteins have been previously shown to be upregulated in several human cancers and to play direct role in cancer progression. Marked upregulation of these proteins, along with their localization and established direct roles in tumor progression, make these molecules promising candidates as drug targets or proteins for sensitive PPGL imaging.
The biochemical phenotype of PGLs is highly dependent on the underlying genetic background and tumor location. PGLs at extra-adrenal locations usually do not express phenylethanolamine N-methyltransferase (PNMT), the enzyme required for epinephrine production, which was explained by the absence of glucocorticoids. PGLs with pathogenic variants in the Harvey rat-sarcoma viral oncogene homolog (HRAS) can occur in or outside the adrenal, but always synthesize epinephrine independently of localization. We characterized the signaling pathways through which pathogenic variants in HRAS influence PNMT expression. We analyzed catecholamines, cortisol, and transcriptional features of PGL tissues with known genetic background. Genetically modified rat pheochromocytoma cells carrying pathogenic variants in Hras were generated and analyzed for regulation of Pnmt expression. Elevated epinephrine contents in PGLs with pathogenic variants in Hras were accompanied by an enrichment in mitogen-activated protein kinase (MAPK) signaling compared with PGLs with pathogenic variants in genes that activate hypoxia pathways. In vitro, Hras pathogenic variants exhibited elevated Pnmt expression and epinephrine biosynthesis through increased phosphorylation of the transcription factor SP1 via MAPK signaling. We provided a molecular mechanism that explains the pathogenic variant-dependent epinephrine production of PGLs.
To understand prognostic immune-cell infiltration signatures in neuroendocrine neoplasms (NENs), particularly PPGL, we analyzed tumor transcriptomic data from The Cancer Genome Atlas (TCGA) and other published tumor transcriptomic data of NENs. We used CIBERSORT to infer immune-cell infiltrations from bulk tumor transcriptomic data from PPGLs, in comparison with gastroenteropancreatic neuroendocrine tumors (GEPNETs) and small-cell lung carcinomas (SCLCs). PPGL's immune signature was validated with NanoString Immune Panel in an independent cohort. We used unsupervised clustering of the immune infiltration scores from CIBERSORT to find immune clusters. Prognostic immune score models for PPGLs and the other NENs were calculated as a linear combination of the estimated infiltration of activated CD8+/CD4+ T cells, activated NK cells, and M0 and M2 macrophages. In PPGLs, we found five dominant immune clusters, associated with M2 macrophages, monocytes, activated NK cells, M0 macrophages and regulatory T cells, and CD8+/CD4+ T cells respectively. Non-metastatic tumors were associated with activated NK cells and metastatic tumors were associated with M0 macrophages and regulatory T cells. In GEPNETs and SCLCs, M0 macrophages and regulatory T cells were associated with unfavorable outcomes and features such as metastasis and high-grade tumors. The prognostic immune-score model for PPGLs and the NENs could predict non-aggressive and non-metastatic diseases. In PPGLs, the immune score was also an independent predictor of metastasis-free survival in a multivariate Cox regression analysis. We concluded that the transcriptomic immune signature in PPGLs correlates with clinical features such as metastasis and prognosis.
The mechanisms triggering metastasis in PPGLs are unknown, hindering therapeutic options for patients with metastatic tumors (mPPGL). By genomic profiling of a large cohort of mPPGLs, we showed that high mutational load, microsatellite instability, and somatic copy-number alteration burden are associated with ATRX/TERT gene mutations and are suitable prognostic markers. Transcriptomic analysis defines the signaling networks involved in the acquisition of metastatic competence and establishes a gene signature related to mPPGLs, highlighting CDK1 as an additional mPPGL marker. Immunogenomics accompanied by immunohistochemistry identifies a heterogeneous ecosystem at the tumor microenvironment level, linked to the genomic subtype and tumor behavior. Specifically, we defined a general immunosuppressive microenvironment in mPPGLs, the exception being PD-L1–expressing MAML3 (a transcription co-activator gene)–related tumors. Our study revealed canonical markers for risk of metastasis and suggests the usefulness of including immune parameters in clinical management for PPGL prognostication and identification of patients who might benefit from immunotherapy.
Therapeutic aspects of pheochromocytoma and paraganglioma
PPGLs with pathogenic mutations in the succinate dehydrogenase (SDHx) subunit B (SDHB) are associated with a high metastatic risk. Somatostatin receptor 2 (SSTR2)–dependent imaging is the most sensitive imaging modality for SDHB–related PPGLs, suggesting that SSTR2 expression is a significant cell-surface therapeutic biomarker of such tumors. We explored the relationship between SSTR2 immunoreactivity and SDHB immunoreactivity, mutational status, and the clinical behavior of PPGLs and evaluated SSTR–based therapies in metastatic PPGLs. We conducted a retrospective analysis of a multicenter cohort of PPGLs at six specialized Endocrine Tumor Centers in Germany, The Netherlands, and Switzerland. Patients with PPGLs participating in the ENSAT registry were included. Clinical data were extracted from medical records, and immunohistochemistry (IHC) for SDHB and SSTR2 was performed in patients with available tumor tissue. Immunoreactivity of SSTR2 was investigated using Volante scores. The main outcome measure was the association of SSTR2 IHC positivity with genetic and clinical-pathological features of PPGLs. Of 202 patients with PPGLs, 50% were SSTR2–positive. SSTR2 positivity was significantly associated with SDHB– and SDHx–related PPGLs, with the strongest SSTR2 staining intensity in SDHB–related PPGLs. Moreover, SSTR2 expression was significantly associated with metastatic disease independent of SDHB/SDHx mutation status. In metastatic PPGLs, the disease control rate with first-line SSTR–based radionuclide therapy was 67% (n = 22, n = 11 SDHx), and with first-line “cold” somatostatin analogs 100% (n = 6, n = 3 SDHx). We concluded that SSTR2 expression was independently associated with SDHB/SDHx mutations and metastatic disease, and we confirmed a high disease-control rate of somatostatin receptor–based therapies in metastatic PPGLs.
Animal model of pheochromocytoma and cell culture studies
Cancer immunotherapy has shown remarkable clinical progress in recent years. Although age is one of the biggest leading risk factors for cancer development, and older adults represent a majority of cancer patients, only a few new cancer immunotherapeutic interventions have been preclinically tested in aged animals. Thus, the lack of preclinical studies focused on age-dependent effect during cancer immunotherapy could lead to different therapeutic outcomes in young and aged animals and to future modifications of human clinical trials. We compared the efficacy of previously developed and tested intra-tumoral immunotherapy, based on the combination of polysaccharide mannan, toll-like receptor ligands, and anti-CD40 (CD40 is a transmembrane protein on antigen-presenting cells) antibody (MBTA immunotherapy), in young (six weeks) and aged (71 weeks) mice bearing experimental PHEO. The results point out that, despite faster growth of PHEO in aged mice, MBTA intra-tumoral immunotherapy is an effective approach independent of age and could be one of the possible therapeutic interventions to enhance immune response to PHEO and perhaps other tumor types in aged and young hosts.
The study was conducted in collaboration with NCI scientists. Autologous tumor cell–based vaccines (ATVs) aim to prevent and treat tumor metastasis by activating patient-specific tumor antigens to induce immune memory. However, their clinical efficacy is limited. Mannan-BAM (MB), a pathogen-associated molecular pattern (PAMP), can coordinate an innate immune response that recognizes and eliminates mannan-BAM–labeled tumor cells. Toll-like receptor (TLR) agonists and anti-CD40 antibodies (TA) can enhance the immune response by activating antigen-presenting cells (APCs) to present tumor antigens to the adaptive immune system. We investigated the efficacy and mechanism of action of rWTC–MBTA, an autologous whole tumor cell vaccine consisting of irradiated tumor cells (rWTC) pulsed with mannan-BAM, TLR agonists, and anti-CD40 antibody (MBTA), in preventing tumor metastasis in several animal models. The efficacy of the rWTC–MBTA vaccine was evaluated in mice using breast (4T1) and melanoma (B16-F10) tumor models via subcutaneous and intravenous injection of tumor cells to induce metastasis. The vaccine's effect was also assessed in a post-operative breast-tumor model (4T1) and tested in autologous and allogeneic syngeneic breast tumor models (4T1 and EMT6). Mechanistic investigations included immunohistochemistry, immunophenotyping analysis, ELISA, tumor-specific cytotoxicity testing, and T cell–depletion experiments. Biochemistry testing and histopathology of major tissues in vaccinated mice were also evaluated for potential systemic toxicity of the vaccine. The rWTC–MBTA vaccine effectively prevented metastasis and inhibited tumor growth in breast tumor and melanoma metastatic animal models. It also prevented tumor metastasis and prolonged survival in the post-operative breast tumor animal model. Cross-vaccination experiments revealed that the rWTC–MBTA vaccine prevented autologous tumor growth, but not allogeneic tumor growth. Mechanistic data demonstrated that the vaccine increased the percentage of antigen-presenting cells, induced effector and central memory cells, and enhanced CD4+ and CD8+ T cell responses. T cells obtained from mice that were vaccinated displayed tumor-specific cytotoxicity, as shown by enhanced tumor-cell killing in co-culture experiments, accompanied by increased levels of Granzyme B, TNF-α, IFN-γ, and CD107a in T cells. T cell–depletion experiments showed that the vaccine's anti-tumor efficacy depended on T cells, especially CD4+ T cells. Biochemistry testing and histopathology of major tissues in vaccinated mice revealed negligible systemic toxicity of the vaccine. We concluded that the rWTC–MBTA vaccine demonstrated efficacy in multiple animal models through T cell–mediated cytotoxicity and has potential as a therapeutic option for preventing and treating tumor metastasis with minimal systemic toxicity.
Publications
- Clinical consensus guideline on the management of phaeochromocytoma and paraganglioma in patients harbouring germline SDHD pathogenic variants. Lancet Diabetes Endocrinol 2023 11:345–361
- Prediction of metastatic pheochromocytoma and paraganglioma: a machine learning modelling study using data from a cross-sectional cohort. Lancet Digit Health 2023 5:e555–e559
- Genomic and immune landscape of metastatic pheochromocytoma and paraganglioma. Nat Commun 2023 14:1122–1142
- Immune signature of pheochromocytoma and paraganglioma in context of neuroendocrine neoplasms associated with prognosis. Endocrine 2023 79:171–179
- Identification of potential molecular targets for the treatment of cluster 1 human pheochromocytoma and paraganglioma via comprehensive proteomic characterization. Clin Proteomics 2023 20:39–55
Collaborators
- James Bibb, PhD, University of Alabama Comprehensive Cancer Center, Birmingham, AL
- Clara C. Chen, MD, Nuclear Medicine Department, Clinical Center, NIH, Bethesda, MD
- Jaydira Del Rivero, MD, Developmental Therapeutics Branch, NCI, Bethesda, MD
- Graeme Eisenhofer, PhD, Universität Dresden, Dresden, Germany
- Hans Ghayee, DO, University of Florida, Gainesville, FL
- Peter Herscovitch, MD, PET Department, Clinical Center, NIH, Bethesda, MD
- Mickey Kuo, MD, Medical Genetics Branch, NHGRI, Bethesda, MD
- Jacques Lenders, MD, Radboud University Medical Centre, Nijmegen, Netherlands
- Frank I. Lin, MD, Molecular Imaging Program, NCI, Bethesda, MD
- W. Marston Linehan, MD, Urologic Oncology Branch, NCI, Bethesda, MD
- Alexander Ling, MD, Department of Radiology, Clinical Center, NIH, Bethesda, MD
- Corina Millo, MD, PET Department, Clinical Center, NIH, Bethesda, MD
- Jirí Neužil, PhD, Institute of Biotechnology, Czech Academy of Sciences, Prague, Czech Republic
- Naris Nilubol, MD, FACS, Surgical Oncology Program, NCI, Bethesda, MD
- Svenja Nolting, MD, PhD, Ludwig-Maximilians-Universität Klinikum, München, Germany; Universitätsspital Zürich, Zürich, Switzerland
- Christina Pamporaki, MD, Universitätsklinikum Carl Gustav Carus, Technische Universität Dresden, Dresden, Germany
- Jiri Petrak, PhD, Institute of Biotechnology, Czech Academy of Sciences, Prague, Czech Republic
- Ondrej Petrak, MD, PhD, General University Hospital, Prague, Czech Republic
- Margarita Raygada, PhD, Section on Endocrinology and Genetics, NICHD, Bethesda, MD
- Mercedes Robledo, PhD, Spanish National Cancer Centre (CNIO), Madrid; Centro de Investigación Biomédica en Red de Enfermedades Raras (CIBERER), Madrid, Spain
- Jared Rosenblum, MD, Neuro-Oncology Branch, NCI, Bethesda, MD
- Douglas Rosing, MD, Translational Medicine Branch, NHLBI, Bethesda, MD
- Kelly Roszko, MD, PhD, Skeletal Disorders and Mineral Homeostasis Section, NIDCR, Bethesda, MD
- Babak Saboury, MD, Nuclear Medicine Department, CC, NIH, Bethesda MD
- David Taieb, MD, PhD, Aix-Marseille Université, Hôpital Universitaire de La Timone, Marseille, France
- Henri Timmers, MD, PhD, Radboud University Medical Centre, Nijmegen, Netherlands
- Arthur S. Tischler, MD, PhD, New England Medical Center, Boston, MA
- Richard Tothill, PhD, University of Melbourne Centre for Cancer Research, Melbourne, Australia
- Brad Wood, MD, PhD, Radiology Department, Clinical Center, NIH, Bethesda, MD
- Chunzhang Yang, PhD, Neuro-Oncology Branch, NCI, Bethesda, MD
- Deena Zeltser, MD, Office of the Clinical Director, NICHD, Bethesda, MD
- Zhengping Zhuang, MD, PhD, Neuro-Oncology Branch, NCI, Bethesda, MD
Contact
For more information, email karel@mail.nih.gov.