
Mechanisms Regulating Fate and Maturation of Forebrain GABAergic Interneurons

- Timothy J. Petros,
PhD, Head, Section on Cellular and Molecular Neurodevelopment - Yajun Zhang, BM, Biologist
- Abhilash Appu, PhD, Staff Scientist
- Arushi Chauhan, PhD, Postdoctoral Fellow
- Jianing Li, PhD, Postdoctoral Fellow
- Matthew Manion, PhD, Postdoctoral Fellow
- Vedant Garg, BS, Postbaccalaureate Fellow
- Ane Martiartu, BS, Postbaccalaureate Fellow
- Anthony Tanzillo, BS, Postbaccalaureate Fellow
Proper brain function requires a balance between excitatory projection neurons and GABAergic inhibitory interneurons. Although interneurons constitute the minority (about 20%) of neurons in the brain, they are the primary source of inhibition and are critical components in the modulation and refinement of the flow of information throughout the nervous system. Interneurons are an extremely heterogeneous cell population, with distinct morphologies, connectivity, neurochemical markers, and electrophysiological properties. This incredible diversity and heterogeneity of interneurons was observed over a century ago, with Ramón y Cajal hypothesizing in Recollections of My Life that “The functional superiority of the human brain is intimately linked up with the prodigious abundance and unaccustomed wealth of the so-called neurons with short axons.” Abnormal development and function of interneurons has been linked to the pathobiology of numerous neurological and psychiatric diseases, such as epilepsy, schizophrenia, and autism. Many genes implicated in brain disorders are enriched in young interneurons, and thus a thorough description of the cellular and molecular mechanisms regulating this diverse cell population is necessary to understand both normal development and disease models.
The lab focuses on understanding how intrinsic genetic and epigenetic programs interact with the local brain environment to generate this incredible diversity of interneuron subtypes during normal development and in disease models (Figure 1). We take a multifaceted approach to this issue, utilizing both in vitro and in vivo approaches to identify candidate mechanisms that regulate interneuron fate decisions. We strive to develop cutting-edge techniques that will overcome the many challenges of studying interneuron development. Our ultimate goal is to discover genetic cascades and signaling mechanisms that direct interneuron differentiation and maturation during normal development and in disease states.
Figure 1. Schematic summarizing interneuron development and our studies
Interneuron progenitors (green) originate from the ventral telencephalon during embryogenesis and migrate tangentially to populate all regions of the adult forebrain. Understanding the fate, maturation, and function of these cells is critical to understand both normal brain development and mechanisms underlying neurodevelopmental disorders.
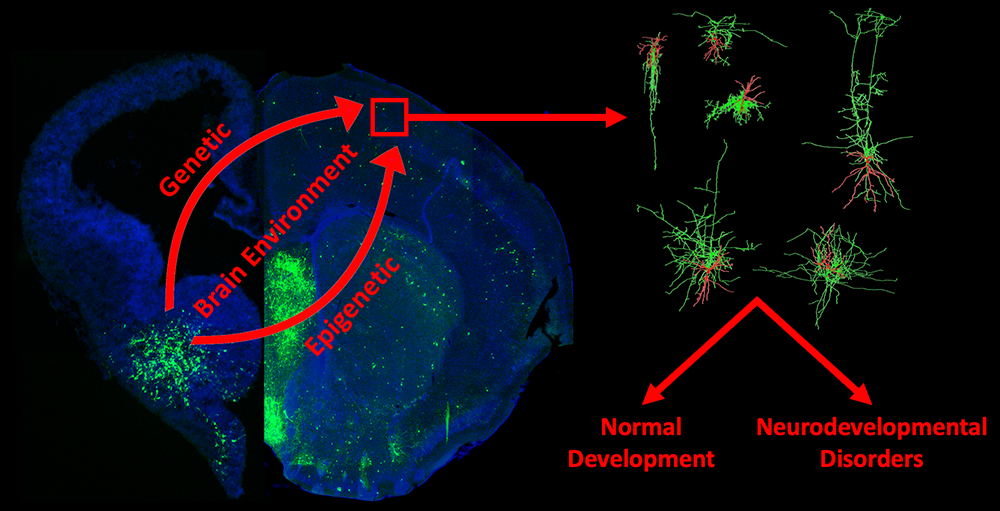
Figure 1. Schematic summarizing interneuron development and our studies
Interneuron progenitors (green) originate from the ventral telencephalon during embryogenesis and migrate tangentially to populate all regions of the adult forebrain. Understanding the fate, maturation, and function of these cells is critical to understand both normal brain development and mechanisms underlying neurodevelopmental disorders.
Reducing methylation of histone 3.3 lysine 4 in the medial ganglionic eminence and hypothalamus recapitulates neurodevelopmental disorder phenotypes.
Methylation of lysine 4 on histone H3 (H3K4) is enriched on active promoters and enhancers and correlates with gene activation. Disruption of H3K4 methylation is associated with numerous neurodevelopmental diseases (NDDs), autism spectrum disorder, schizophrenia, and substance-related disorders (Figure 2A). Two common symptoms of many H3K4–related disorders are intellectual disability and abnormal body growth. Studying H3K4 methylation has been challenging because H3K4 can be methylated by six different proteins, and experimentally deleting a single methyltransferase can lead to confounding compensations by other proteins. To overcome this challenge, we utilized a Cre–dependent transgenic mouse in which the H3K4 lysine is mutated to a methionine (H3K4M), thus blocking methylation at this residue. The homozygous H3K4M mouse contains two wild-type (WT) H3K4 alleles and two mutant H3K4M alleles, acting as a functional heterozygote, and is thus a good model for exploring the role of altered H3K4 methylation in human disease. We generated Nkx2.1-Cre;H3K4M mice to target this mutation to the medial ganglionic eminence (MGE) and the hypothalamus, two brain regions associated with intellectual disability and regulation of body growth (Figure 2B). Such mutant mice have fewer forebrain interneurons, deficient network rhythmogenesis, and increased spontaneous seizures and seizure susceptibility (Figure 2C). Mutant mice are significantly smaller than control littermates, but they eventually became obese because of striking changes in the genetic and cellular hypothalamus environment in these mice (Figure 2D). Perturbation of H3K4 methylation in these cells produces deficits in numerous NDD–associated behaviors, such as increased anxiety and altered memory and sensorimotor responses (Figure 2E). We observed a striking cellular disorganization in the hypothalamus of H3K4M mutant mice, likely compromising the normal blood-brain behavior and leading to numerous hypothalamus-related deficits (Figure 2F). Our single-cell sequencing experiments reveal many transcriptional changes in both the MGE and hypothalamus, which underlie cell-fate and behavioral changes in these mutant mice (Figure 2F). taken together, our findings highlight the critical role of H3K4 methylation in regulating survival and cell-specific gene-regulatory mechanisms in forebrain GABAergic and hypothalamic cells during neurodevelopment to control network excitability and body size homoeostasis. Our article on this study is currently under revision, and a preprint is available [Reference 2].
Figure 2. Disruption of H3K4 methylation in the MGE
A. Role of H3K4 methylation in disease. C. Genetic strategy to express the H3K4M mutation in the MGE and hypothalamus to create a functional heterozygous (Het) mouse. C. Decrease of MGE–derived cortical interneurons in mutant mice. D. K4M homozygous (Hom) mice are smaller in the first postnatal weeks but then become obese from 5–12 months. E. H3K4M mutant mice display increased anxiety and locomotion in open field test. F. Disorganization of astrocytes and tanycytes in the hypothalamus of mutant mice. G. Differentially expressed genes between wild-type (WT) and H3K4M–mutant mice in the embryonic MGE and hypothalamus.
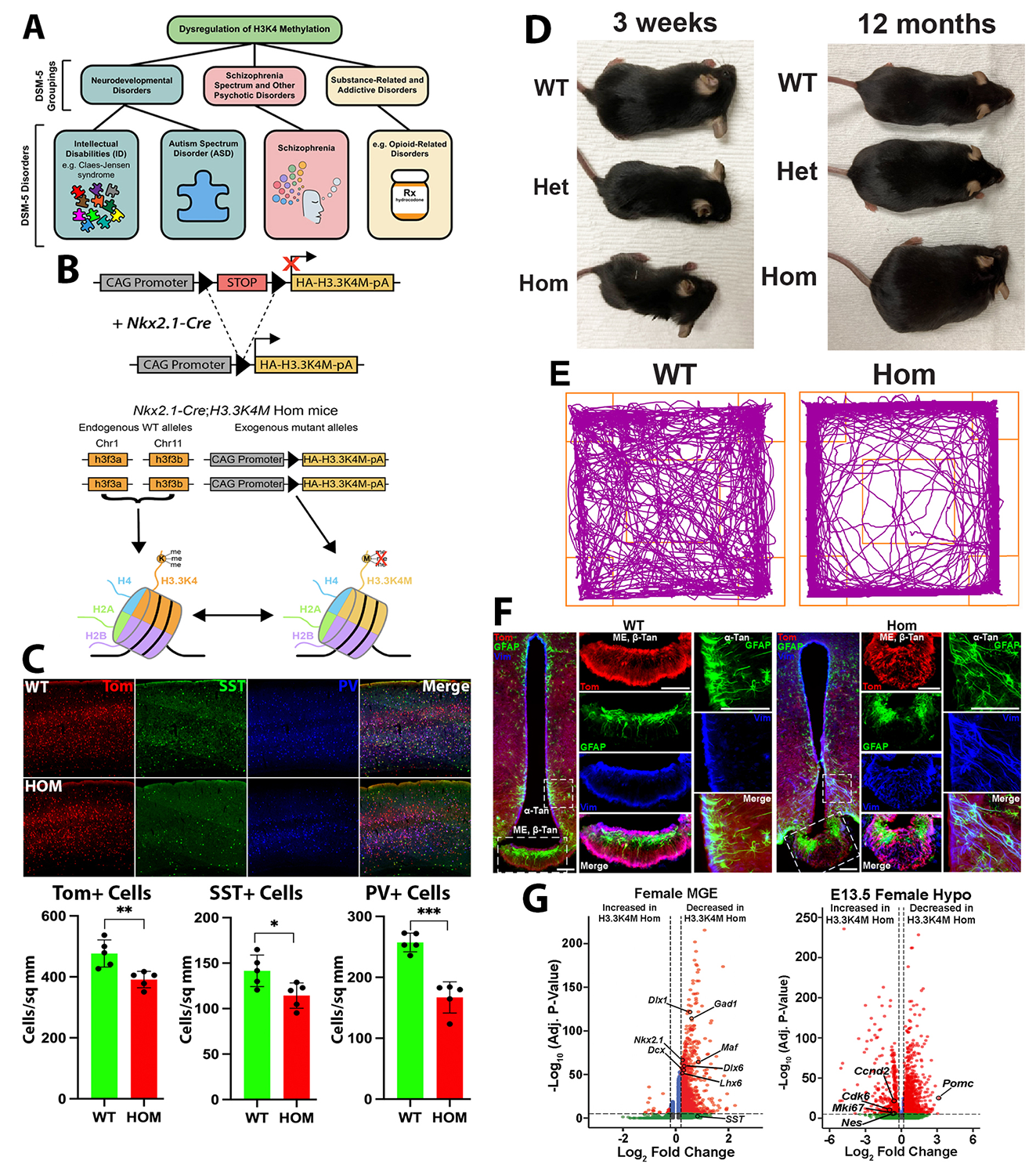
Figure 2. Disruption of H3K4 methylation in the MGE
A. Role of H3K4 methylation in disease. C. Genetic strategy to express the H3K4M mutation in the MGE and hypothalamus to create a functional heterozygous (Het) mouse. C. Decrease of MGE–derived cortical interneurons in mutant mice. D. K4M homozygous (Hom) mice are smaller in the first postnatal weeks but then become obese from 5–12 months. E. H3K4M mutant mice display increased anxiety and locomotion in open field test. F. Disorganization of astrocytes and tanycytes in the hypothalamus of mutant mice. G. Differentially expressed genes between wild-type (WT) and H3K4M–mutant mice in the embryonic MGE and hypothalamus.
Mature interneuron subtypes arise from distinct spatial and temporal subdomains within the caudal ganglionic eminence.
Forebrain inhibitory interneurons are born from transient structures during embryogenesis known as the medial and caudal ganglionic eminences (MGE and CGE, respectively). The MGE and CGE generate distinct, non-overlapping cohorts of interneurons that can be defined by their transcriptomic, morphological, and electrophysiological characteristics. Previous studies transplanted fluorescent embryonic precursors from MGE subdomains into postnatal brains to determine the spatial and temporal origin of mature interneuron subtypes. These studies identified a spatial and temporal organization relating MGE progenitors to mature interneuron subtypes. Combining these insights with gene-expression patterns has generated important insights into MGE development. Surprisingly, whether a similar organization occurs in the CGE has not been explored. The CGE contributes about 30% of interneurons in mice, but humans and primates have a larger proportion of CGE–derived interneurons. Additionally, CGE progenitors are the primary source of tumors and cortical lesions in the tuberous sclerosis complex (TSC), underscoring the importance of understanding development of CGE cells. We harvested fluorescent cells from the anterior, posterior, ventral, and dorsal CGE cells (aCGE, pCGE, vCGE, dCGE) at E13.5 and E15.5 and grafted them into WT neonatal mice cortices (Figure 3A). Brains are harvested 30–35 days post-transplantation, sectioned, and immunostained for CGE–specific interneuron markers (VIP, CCK, Reln, CR, etc.) to correlate spatial and temporal origin with mature CGE–derived interneuron subtypes (Figure 3B). We identified significant spatial biases for most CGE–derived interneuron subtypes to preferentially arise from distinct CGE subdomains (Figure 3C). These biases are relatively stable over time, implying a minimal relationship between temporal birthdate and interneuron subtype (Figure 3D), which differs significantly from the MGE. Our spatiotemporal map of CGE interneuron development (Figure 3E) can now be integrated with datasets showing differential gene expression throughout the CGE to identify candidate genes that regulate fate and maturation of specific CGE–derived interneuron subtypes. This manuscript is currently in revision and a preprint is available at bioRxiv [Reference 3].
Figure 3. Mature interneuron subtypes arise from distinct spatial and temporal subdomains within the caudal ganglionic eminence.
A. Procedure to harvest CGE subdomains from DlxCre-Ai9 mice and transplant these cells into the cortices of neonate WT mice. B. Example of grafted Tom+ cells in P35 brain that have been stained for markers of mature subtypes of CGE–derived interneurons. C. Graphs depicting biased origin of Reelin+ cells arising from the posterior CGE and VIP+ cells arising from the dorsal CGE. D. There is relative stability in production of specific CGE–derived interneurons over time, with only the CR+ population increasing significantly from E13.5 to E15.5. E. Schematic summarizing the spatiotemporal origin of distinct interneuron subtypes within the CGE.
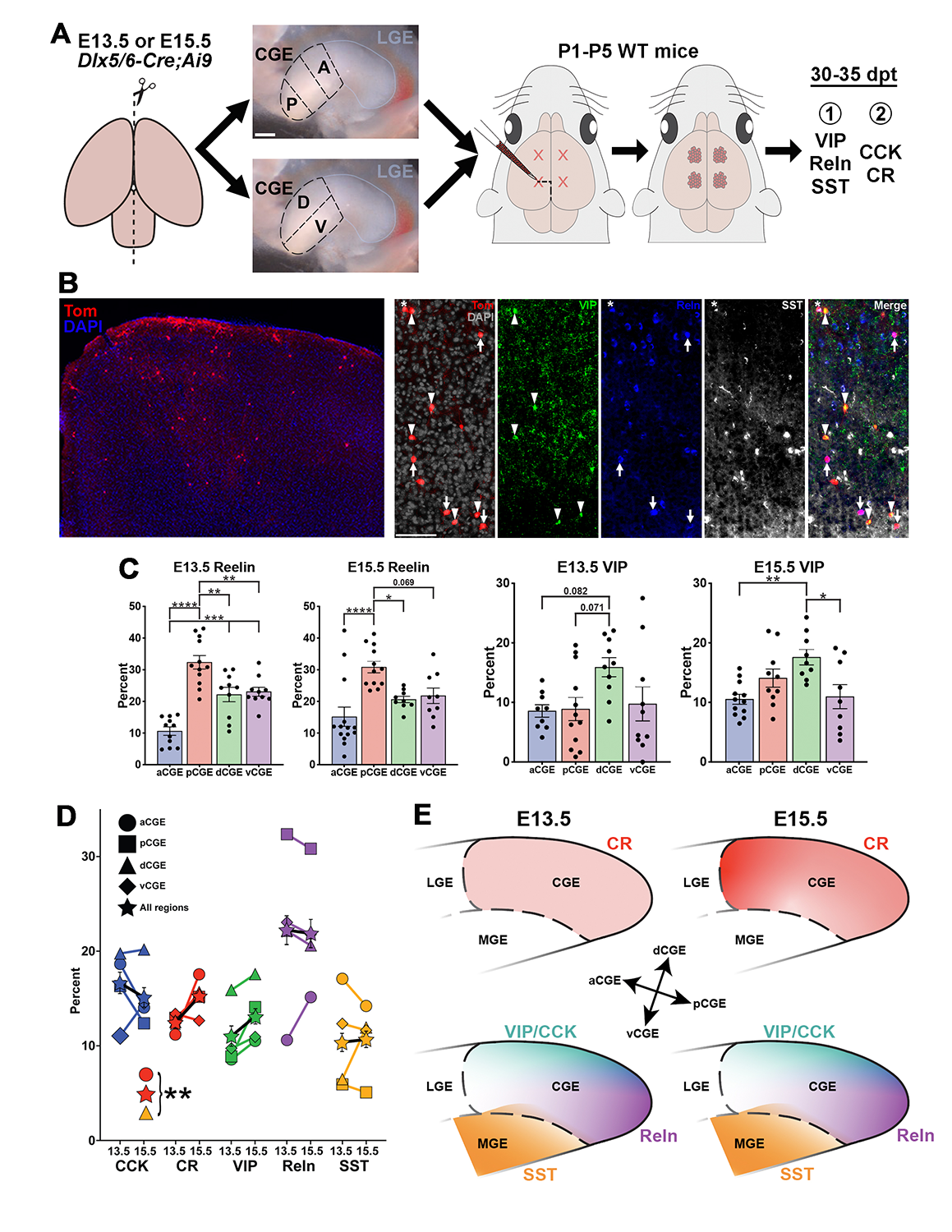
Figure 3. Mature interneuron subtypes arise from distinct spatial and temporal subdomains within the caudal ganglionic eminence.
A. Procedure to harvest CGE subdomains from DlxCre-Ai9 mice and transplant these cells into the cortices of neonate WT mice. B. Example of grafted Tom+ cells in P35 brain that have been stained for markers of mature subtypes of CGE–derived interneurons. C. Graphs depicting biased origin of Reelin+ cells arising from the posterior CGE and VIP+ cells arising from the dorsal CGE. D. There is relative stability in production of specific CGE–derived interneurons over time, with only the CR+ population increasing significantly from E13.5 to E15.5. E. Schematic summarizing the spatiotemporal origin of distinct interneuron subtypes within the CGE.
Perturbing chromatin architecture disrupts gene expression in the MGE.
Characterizing how chromatin architecture regulates proper promoter-enhancer interactions and gene expression is critical to an understanding of normal development and disease etiologies. Many promoter-enhancer interactions are tissue- or cell-type–specific, so that it is critical to study these interactions in vivo. Our previous study identified a direct chromatin interaction between Nkx2.1 (encodes the homeobox protein Nkx-2.1, which regulates genes that play a role in the development and movement of interneurons) and a gene upstream called MAP3K12–binding inhibitory protein 1 (MBIP). Like Nkx2.1, MBIP is also only expressed in the MGE (Figure 4A). Given that Nkx2.1 is a ‘master regulator’ of MGE–derived interneurons, Nkx2.1 represents an ideal locus to explore how brain region–specific chromatin interactions regulate gene expression and cell fate.
We used a cutting-edge genetic strategy to generate a mouse line that disrupts the MGE–specific Nkx2.1-Mbip interaction, as confirmed by Micro-C (Nkx2.1–CTCF mice, Figure 4B). There is a strong reduction of Mbip and a moderate downregulation of Nkx2.1 in the MGE in these mice (Figure 4C). We are currently quantifying the number of SST+ (somatostatin-expressing) and PV+ (fast-spiking parvalbumin-positive) interneurons in the cortex to determine whether there are any changes in the density and/or proportion of MGE–derived interneurons in the Nkx2.1–CTCF mutant mice. Success with this project will motivate us to apply these genetic perturbations to other genomic loci at critical fate-determining genes that have brain region–specific chromatin interactions. We have already generated another mouse line using similar technology to disrupt a different brain region–specific chromatin interaction in order to study its role in interneuron development.
Figure 4. Perturbation of chromatin interaction linking Nkx2.1 and MBIP
A. MGE–specific interaction between Nkx2.1 and Mbip (black arrowheads). B. 6 CTCF sites were inserted between Nkx2-1 and Mbip to block this interaction (top). Micro-C confirmed loss of this Nkx2.1–Mbip interaction in the MGE (black arrowheads) with a corresponding new interaction at the ectopic CTCF sites (purple arrowheads). C. Strong decrease of Mbip mRNA and moderate reduction of Nkx2.1 mRNA in the MGE of Nkx2.1–CTCF mice.
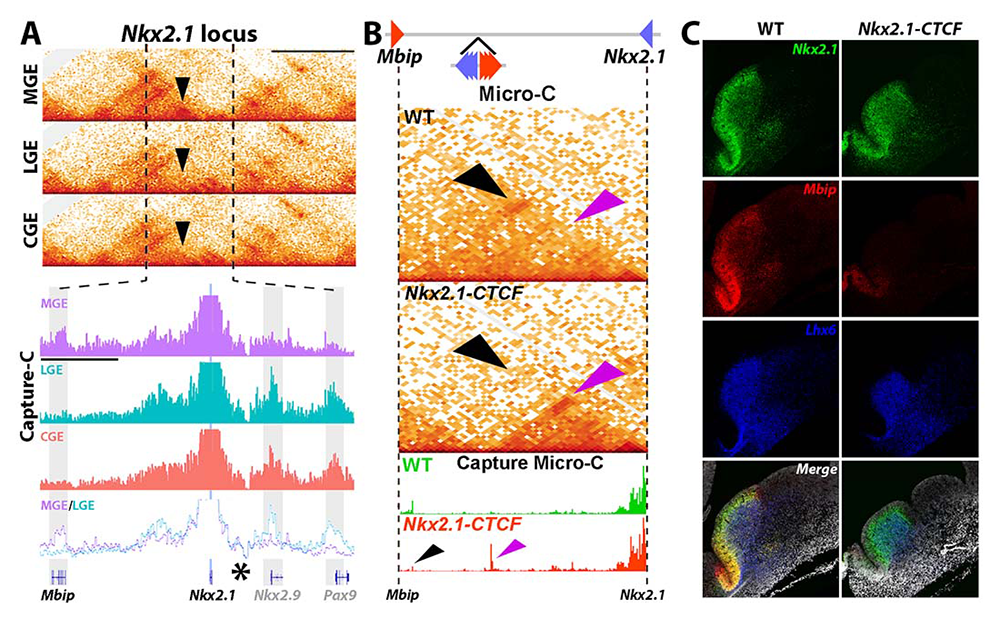
Figure 4. Perturbation of chromatin interaction linking Nkx2.1 and MBIP
A. MGE–specific interaction between Nkx2.1 and Mbip (black arrowheads). B. 6 CTCF sites were inserted between Nkx2-1 and Mbip to block this interaction (top). Micro-C confirmed loss of this Nkx2.1–Mbip interaction in the MGE (black arrowheads) with a corresponding new interaction at the ectopic CTCF sites (purple arrowheads). C. Strong decrease of Mbip mRNA and moderate reduction of Nkx2.1 mRNA in the MGE of Nkx2.1–CTCF mice.
Additional Funding
- NICHD Career Development Award (Postdoctoral Fellow Arushi Chauhan)
- NICHD Career Development Award (Postdoctoral Fellow Jianing Li)
Publications
- Deletion of a single CTCF motif at the boundary of a chromatin domain with three FGF genes disrupts gene expression and embryonic development. Dev Cell 2025 60:1838-1853
- Reducing methylation of histone 3.3 lysine 4 in the medial ganglionic eminence and hypothalamus recapitulates neurodevelopmental disorder phenotypes. bioRxiv 2025 doi:10.1101/2025.05.02.651761;preprint
- Mature interneuron subtypes arise from distinct spatial and temporal subdomains within the caudal ganglionic eminence. bioRxiv 2025 doi:10.1101/2025.07.28.667082;preprint
- Dynamic regulation of cholesterol metabolism genes in dopaminergic neurons following methamphetamine treatment as revealed by single-nucleus RNA sequencing. bioRxiv 2025 doi:10.1101/2025.07.28.667272;preprint
Collaborators
- Susan Amara, PhD, Laboratory of Molecular and Cellular Neurobiology, NIMH, Bethesda, MD
- James Bourne, PhD, Section on Cellular and Cognitive Neurodevelopment, NIMH, Bethesda MD
- Ryan Dale, MS, PhD, Bioinformatics & Scientific Programming Core, NICHD, Bethesda, MD
- Soohyun Lee, PhD, Unit on Functional Neural Circuits, NIMH, Bethesda, MD
- Chris McBain, PhD, Section on Cellular and Synaptic Physiology, NICHD, Bethesda, MD
- Giulia Quattrocolo, PhD, Norwegian University of Science and Technology, Kavli Institute for Neurosciences, Trondheim, Norway
- Pedro Rocha, PhD, Unit on Genome Structure and Regulation, NICHD, Bethesda, MD
- Lorna Role, PhD, Circuits, Synapses and Molecular Signaling Section, NINDS, Bethesda MD
- Mark Stopfer, PhD, Section on Sensory Coding and Neural Ensembles, NICHD, Bethesda, MD
Contact
For more information, email tim.petros@nih.gov or visit https://www.nichd.nih.gov/research/atNICHD/Investigators/petros.