
Cellular and Molecular Mechanisms of Lymphatic Disorders

- Sarah E. Sheppard,
MD, PhD, MS, Head, Unit on Vascular Malformations - Andrea Bowling, NP, Nurse Practitioner
- Deena Zeltser, MD, Staff Clinician
- Christopher Marshall, BA, Research Specialist II
- Luciana Daniela Garlisi Torales, MD, Postdoctoral Fellow
- Lola Zerbib, PharmD PhD, Postdoctoral Fellow
- Jessica Johnson, BA, Predoctoral Fellow (Ox-Cam)
- Kwabena Aboagye, BS, Postbaccalaureate Fellow
- Audrey Breckenridge, BS, Postbaccalaureate Fellow
- Georgia Krikorian, BA, Postbaccalaureate Fellow
- Laila Lawson, BS, Postbaccalaureate Fellow
- Parinita Nautiyal, BA, Postbaccalaureate Fellow
- Ben Sempowski, BS, Postbaccalaureate Fellow
- Rachel Amedume, BS, Howard University Undergraduate Honors Program Biology Student/Postbaccalaureate Fellow
The primary goal of our translational research group is to develop efficacious therapies for patients with complex lymphatic anomalies. To do this, we seek to understand the molecular etiologies of these complex lymphatic malformations, how the molecular etiologies alter molecular signaling, and how this affects the cellular mechanisms regulating normal lymphatic development. Ultimately, these answers will allow us to develop novel therapies.
Complex lymphatic anomaly is a term that encompasses four different complex lymphatic malformations: central conducting lymphatic anomaly (CCLA); generalized lymphatic anomaly (GLA); Kaposiform lymphangiomatosis (KLA); and Gorham Stout disease (GSD). Patients suffer from symptoms such as pleural effusions, pericardial effusions, ascites, and bone lesions, which can cause significant morbidity and even death. Currently, there is only one medication approved for patients with complex lymphatic anomalies caused by PIK3CA, a gene mutation known to cause lymphatic malformations. Similar precision-medicine approaches are needed for patients with other complex lymphatic anomalies.
Research in our lab will combine patient studies and genomics with the zebrafish model to identify novel therapies. The zebrafish model allows us to manipulate the genetics so as to rapidly create patient-based models, image the developing vasculature, understand cellular dynamics in vivo, and perform drug screening.
A bedside-to-bench precision medicine program for lymphatic anomalies
The figure shows a circle with the major components of the research in the lab: a child with abnormal lymphatics, a DNA molecule, a representation of a zebrafish model and organoid model, and a pill bottle. This represents a bedside-to-bench-to-bedside program using organoid and zebrafish to model patients' lymphatic anomalies and to develop therapies that will be translated back to the patients.
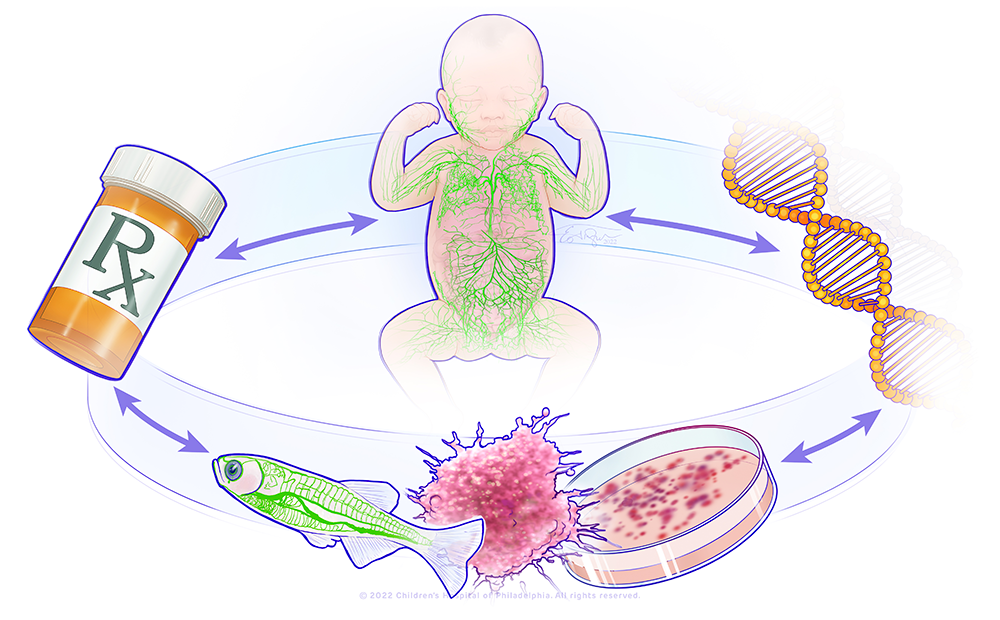
A bedside-to-bench precision medicine program for lymphatic anomalies
The figure shows a circle with the major components of the research in the lab: a child with abnormal lymphatics, a DNA molecule, a representation of a zebrafish model and organoid model, and a pill bottle. This represents a bedside-to-bench-to-bedside program using organoid and zebrafish to model patients' lymphatic anomalies and to develop therapies that will be translated back to the patients.
Natural history study of lymphatic disorders
In 2023, the lab launched a prospective natural history study of individuals with lymphatic anomalies to systematically evaluate the disease phenotypes and long-term outcomes. As these are rare diseases, patients may often face misdiagnosis. Moreover, there is a lack of information about the long-term outcomes. For deep phenotyping, patients are seen by telemedicine by the primary team or at the NIH Clinical Center by the primary team and several specialists. The study will allow us to provide improved prognostication to families, establish screening/monitoring guidelines, determine best practices for genetic diagnosis, and explore fertility outcomes for those on long-term medication management. The study will allow us to identify novel end-points for future clinical trials. To date, the protocol has enrolled 94 participants with primary lymphatic anomalies.
Using genetic diagnostics to provide diagnoses and expand the phenotype of vascular malformations
Vascular malformations cause disability; identification of novel features of disease is essential for disease surveillance.
Verrucous venous malformation (VVM) is frequently misdiagnosed. An individual with this condition demonstrated the value of genetic testing to inform correct diagnosis. Consent was provided for retrospective chart review and publication. At birth, the affected individual had a red flat patch with overgrowth of his right hand, arm, and shoulder. The overgrowth and red color persisted and began to cause pain, which prompted his initial evaluation at age 10 years, leading to a diagnosis of a capillary vascular malformation and overgrowth. Routine magnetic resonance imaging of the right arm showed no definitive vascular anomaly. At 12 years-of-age, hyperkeratosis prompted a biopsy for diagnosis. Pathology from two skin biopsies was most consistent with a capillary malformation and showed mildly acanthotic epidermis with hyperkeratosis overlying dilated, thin-walled vascular channels. We performed a 34–gene somatic genetic testing panel on the biopsy tissue. Results showed a likely pathogenic variant in the MAP3K3 gene c.674C>T, p.Ser225Phe at variant allele fraction of 4.2%–4.6% from sample 1 and 7.3%–8.0% from sample 2, confirming the diagnosis of VVM. Somatic genetic testing provided this affected individual with the diagnosis of VVM when radiology, pathology, and clinical findings were discrepant. This new genetic diagnosis influences treatment and prognosis.
Some vascular anomalies, such as hamartomas associated with PTEN hamartoma tumor syndrome (PHTS) and fibroadipose vascular anomaly (FAVA, often due to PI3KCA variants), share similar clinical, radiological, and histopathological presentations that are a challenge for clinicians to provide an accurate diagnosis. Genetic testing can help clinicians differentiate between these two vascular anomalies to provide proper treatment for patients. An 11-year-old female with macrocephaly presented with a painful lesion in her right ankle and was initially diagnosed with FAVA and treated with the immunosuppressant sirolimus. Initial genetic testing from a biopsy sample was negative. Subsequently, however, repeat clinical genetic testing and deep exome sequencing from a second tissue biopsy sample identified a mosaic variant in PTEN (NM_00314.7) c.683delA p.Asn228Ilefs*28 with a variant allele fraction (VAF) of 2.0%–2.1%, ultimately changing the diagnosis from FAVA to a PTEN hamartoma. To evaluate the germline status of this patient, PTEN sequencing and deletion duplication testing from saliva identified a different variant in PTEN(NM_000314.4) c.202_209+18delins27, estimated to be 20%–30% VAF. Sanger sequencing validated this novel variant as germline, leading to cancer screening in the patient. This case exemplifies the need for genetic reevaluation, as sequencing technology continues to update rapidly, and for repeat sampling in cases of suspected mosaicism, and it supports the two-hit hypothesis in the development of vascular malformations. It also emphasizes the importance of genetic diagnosis in vascular malformations, especially in this case, which led to the identification of a cancer-predisposition syndrome.
Therapies for KRAS–related lymphatic disorders
Previously, we found that activating variants in the KRAS gene cause complex lymphatic anomalies. KRAS is a RAS-GTPase, and pathogenic variants are known to drive cancers as well as developmental disorders. Previously, we showed that KRAS variants can drive lymphatic malformations in the zebrafish, which can be treated with MEK (a kinase that phosphorylates mitogen-activated protein kinase) inhibitors.
However, issues regarding therapies for KRAS–related lymphatic anomalies still remain. First, different variants in KRAS lead to up-regulation of MAPK signaling (a central signaling pathway) by different mechanisms. Second, the patients have different phenotypes. Third, previous patients with KRAS–related lymphatic anomalies have mixed responses to MEK inhibitors. Therefore, we developed new zebrafish models for three different KRAS–related variants that have lymphatic defects. Currently, these models are being used for drug-therapy screening.
Cellular and molecular mechanisms of lymphatic malformations
Novel candidate genes and known genetic causes associated with disease must be validated to ensure that they truly cause disease. Mechanisms of disease must be elucidated for known causes of lymphatic disease as a first step toward identifying a precision-based treatment. Previously, we identified pathogenic variants in the RIT1 gene (RIT1 belongs to the Ras family of GTPases) as a novel cause of CCLA [Liu M et al., Eur J Hum Genet 2022;30:1022]. However, the mechanisms that drive disease are unknown. Therefore, we created a mosaic zebrafish model of RIT1 lymphatic anomalies and a zebrafish knock-in with a patient allele to evaluate the lymphatic anomalies, so as to determine the effect on the cellular and molecular mechanisms driving lymphatic development, and, in the future, to identify therapies. The group is currently evaluating the phenotypes as well as potential therapies in these models.
Publications
- Multiple genomic technologies validate rare novel variant and direct medical care in vascular anomalies. Am J Med Genet A 2025 e64126
- Somatic genetic testing provides diagnosis of verrucous venous malformation in an individual with discrepant radiology, pathology, and clinical findings. J Vasc Anom (Phila) 2025 6(4):e120
- A pilot study to evaluate neurodevelopmental outcomes in a pediatric cohort with genodermatoses. Am J Med Genet A 2025 197(7):e63989
- Workforce shortage of pediatric dermatologists: a medical student's perspective. Cutis 2025 115(5):E18-E20
- Expansion of the phenotype of lymphatic anomalies caused by somatic activating BRAF variant. Pediatr Blood Cancer 2025 72(11):e32015
Collaborators
- Maureen Cetera, PhD, University of Minnesota, Minneapolis, MN
Contact
For more information, email sarah.sheppard@nih.gov or visit https://www.nichd.nih.gov/research/atNICHD/Investigators/sheppard.