
Membrane Pathophysiology of the Infectious Diseases Influenza and Toxoplasmosis

- Joshua Zimmerberg,
MD, PhD, Head, Section on Integrative Biophysics - Paul S. Blank, PhD, Staff Scientist
- Svetlana Glushakova, MD, PhD, Staff Scientist
- Irene Jimenez Munguia, PhD, Visiting Fellow
- Avijit Sardar, PhD, Visiting Fellow
- Melanie Brunet Torres, PhD, Postdoctoral Intramural Research Training Award Fellow
- Hang Waters, MS, Biologist
- Elena Mekhedov, MA, Contractor
- Glen Humphrey, PhD, Guest Researcher
- Phuong (Audrey) Pham, BS, Postbaccalaureate Fellow
Eukaryotic life must create the many shapes and sizes of the system of internal membranes and organelles that inhabit the variety of cells in nature, i.e., membranes that must remodel for cells to repair damaged plasmalemma and deal with infectious agents such as viruses and parasites. Such basic membrane mechanisms must be highly regulated and highly organized in various hierarchies in space and time to allow the organism to thrive despite environmental challenges, genetic instability, unpredictable food supply, and physical trauma. We are using the expertise and techniques we perfected over the years to address various biological problems that have in common the underlying regulation or disturbance of protein/lipid interactions. The overall goal of this project is to determine the physico-chemical mechanisms of membrane remodeling in cells, and how the knowledge of these mechanisms can lead to the amelioration of disease, particularly muscular dystrophies where muscle weakness becomes debilitating.
Even in a non-pandemic year, the influenza A virus (IAV) causes significant morbidity and mortality in human populations. HIV and Sars-Cov-2, like IAV, cause two of top five pandemic killers; all three have similar membrane-active components that govern the assembly of these viruses and the membrane fusion of their entry to infect. Clearly, a deep understanding of their common mechanisms may identify vulnerabilities for therapeutic or immunogenic attack.
There is a parasite whose approximate infection rate is up to one third of the global population: Toxoplasma. The causative parasite (T. gondii) passes from the mother to the fetus, and causes congenital toxoplasmosis, a considerable cause of morbidity and mortality throughout the world, especially in developing countries. One estimate of the incidence of congenital toxoplasmosis is 190,000 cases annually, a rate of about 1.5 cases of congenital toxoplasmosis per every 1000 live births. But if an infection is acquired early in pregnancy and treatment is not given, severe consequences, such as fetal death, are likely. Unlike bacteria, eukaryotic parasites such as T. gondii share more proteins with the human host, and we must always be prepared to find molecular targets for drug-resistant strain emergence, or vaccine candidates that are unique to the parasite. This year, we report the development of new tools leading to the discovery of a novel pathophysiological membrane process in the invasion of host cells by T. gondii, which paves the way to the identification of a completely unique set of molecules that may be good candidates for drug discovery.
The invasion pore is caused by rhoptry exocytosis.
Invasion of host cells is a critical step in the lytic cycle of the protozoan parasite Toxoplasma gondii and is essential for establishing infection. The overall process of invasion is well conserved among apicomplexan parasites, including T. gondii and the related parasites that cause malaria (Plasmodium spp.) and cryptosporidiosis (Cryptosporidium spp.). Invasion involves the recognition of and attachment to a host cell, delivery of parasite proteins into that host cell, and parasite-driven internalization. Prior to invasion, the proteins to be translocated into the host cell are stored in the rhoptries, which are club-shaped exocytic organelles consisting of a posterior bulb and an elongated neck docked at the apical end of the parasite. Delivery of rhoptry proteins into the host cell is essential for invasion and crucial for parasite virulence. For example, translocated rhoptry neck protein2 (RON2) forms a complex at the host-cell plasma membrane, which serves as a binding site for the parasite, providing traction at the “moving junction” through which the parasite propels itself into the host cell during invasion. Translocated rhoptry bulb proteins (ROPs) act downstream of invasion to suppress the innate immune response and manipulate the host cell in ways that facilitate parasite intracellular survival and replication.
The detailed structures of the rhoptry secretory apparatus (RSA) and many of its molecular components have been determined in Toxoplasma, Plasmodium, and Cryptosporidium. The RSA lies at the extreme apical tip of the parasite plasma membrane and forms the apical rosette, a transmembrane structure to which an underlying membrane-bound apical vesicle (AV) is typically docked. Exocytosis of lumenal rhoptry proteins through the rosette therefore involves two membrane fusion events: the rhoptry tip with the AV, and the AV with the parasite plasma membrane. Despite this knowledge, the mechanism by which the exocytosed rhoptry proteins are translocated into the host cell remains unknown, and is one of the most enigmatic of the molecular events during T. gondii invasion, given the appearance of rhoptry proteins in the host-cell cytoplasm during invasion. The presence of rhoptry proteins in the host-cell cytoplasm appears to violate a fundamental cell-biological law governing conservation of membrane and secretory protein topology, i.e., that protein domains co-translationally inserted into the lumen of the ER remain lumenal throughout the secretory pathway, including within secretory granules, and once released from those granules face the extracellular space. Accordingly, the lumen of the rhoptry is topologically equivalent to the extracellular space and not to the host-cell cytoplasm. Proteins released from the lumen of the rhoptry can change their topology only by traversing a limiting membrane such as the host-cell plasma membrane. Thus, fusion between the parasite and host-cell plasma membrane is ruled out as a mechanism of translocation, because the exocytosed rhoptry proteins would still remain outside the host cell (indeed the word “exocytosis” is the conjunction of two Greek roots exo=outside and cyto=cell). Fusion of the parasite and host-cell plasma membranes is also ruled out experimentally, given that our earliest electrophysiology experiments showed that host-cell plasma membrane capacitance, which measures membrane surface area, does not increase during invasion [Suss-Toby E et al., Proc Natl Acad Sci USA 1996;93:8413], as would be expected if the two membranes were to fuse.
In this brief synopsis of our papers, we describe the detailed analysis of the electrophysiological characteristics of the invasion-associated change in host-cell plasma membrane conductance, leading to the discovery of the invasion pore and the role of rhoptry exocytosis in the invasion poration process. We developed a new high-speed fluorescence microscopy–based assay that allows us to spatially monitor the plasma-membrane barrier integrity as the change in cytosolic chemistry as extracellular ions enter [Reference 3]. This assay confirmed that, immediately preceding invasion, the parasite creates a real transient perforation in the host-cell membrane and is not merely an electrical phenomenon. We also demonstrated that the perforation occurs at the point on the host-cell membrane in contact with the interacting parasite’s apical end.
The advantage of a fluorescent assay is that mutants can be screened more rapidly, and therapeutics can be developed using drug libraries. We used this assay to test whether parasites conditionally depleted of five different proteins that function in rhoptry exocytosis are able to induce the perforation. In all cases, a block in rhoptry exocytosis led to a block in host cell perforation. These results are consistent with a model in which material stored within the rhoptries is exocytosed upon contact with the host cell, causing a transient perforation in the host-cell membrane through which rhoptry effector proteins are delivered. One of these mutants (i.e., a mutant lacking RASP2, the rhoptry apical surface protein 2, a protein regulating the rhoptry protein discharge) blocks perforation, even though the RSA signaling is intact, showing that rhoptry secretion itself is needed for poration, and is not a consequence of the receptor-ligand binding between the parasite and the host cell prior to exocytosis leading to RSA activation. We tested cells directly by electrophysiology and found no electrical evidence for cell perforation when RASP2 is inactivated. Therefore, rhoptry exocytosis is essential for the host-cell poration.
The rhoptry neck protein RON2, described above, is the only known component of the moving junction that is inserted as a transmembrane protein into the host-cell plasma membrane. It was thus reasonable to hypothesize that the reason that the RASP2 mutant blocked poration was that RON2 was not delivered to the host-cell membrane. We tested this hypothesis by measuring host-cell plasma membrane conductance and calcium entry during T. gondii invasion in parasites with and without RON2. Transients were collected with the T. gondii wild-type (WT) RH strain (widely used in Toxoplasma-related research, classified as highly virulent) using high-bandwidth direct current measurements of host cells (COS1) under voltage-clamp conditions in the whole-cell configuration [Reference 4]. Simultaneously, parasite invasion was monitored by DIC (differential interference contrast) microscopy. Several parasites were delivered to the host cells to increase the probability of capturing many invasions and the associated transient increases in whole-cell current. Invading tachyzoites (the quickly dividing stages of Toxoplasma gondii inside parasitophorous vacuoles) showed a clear constriction, indicative of parasite entry into the host cell, and a single large current change was detected, always prior to the constriction of each parasite; many transients were recorded when many parasites invaded the same host cell. We observed no instance of invasion without a transient. Analysis revealed that every transient possessed characteristic waveform features, i.e., a fast rise to a peak and a slower fall to a new baseline. No obvious difference in transient conductance was seen based on the order of parasite entry, i.e., the first, middle, and last transients seemed similar. Thus, each invading WT T. gondii tachyzoite produces an individual, independent conductance transient.
To investigate whether RON2 insertion (and thus “complete” moving junction formation) is associated with the appearance of transients, we used both current recording experiments and the calcium influx assay to independently interrogate the ability of RON2 knockdown (KD-RON2) parasites to generate the conductance and calcium transients. As seen in both assays, KD-RON2 parasites generated conductance and calcium transients at levels similar to those seen in WT parasites, which stands in stark contrast to their near absence when the parasites are depleted of proteins that regulate rhoptry exocytosis. Together, the conductance and calcium transient results rule out the hypothesis that RON2 insertion into the host-cell plasma membrane is itself the poration process. Therefore, the transient conductance increase induced by WT parasites occurs independently of complete moving junction formation.
Parameterization of the transients was consistent with similar pores produced by WT and KD-RON2 parasites, but a longer peak conductance duration was observed in the averaged transients and confirmed in the analysis of the individual peak durations. The initial analysis of the time course of conductance during each transient revealed step-like transitions in both the rising and falling phases of the transients. To analyze the magnitudes and numbers of the rapid, step-like changes in conductance, each transient was processed using piecewise constant (PWC) signal denoising. From the resulting conductance values, the density distribution of the differences in adjacent conductance levels was obtained. WT parasite–induced transients display a probability density peak at 0.26 nS, which can be considered a characteristic quantal step size. We tested this hypothesis further by both parametric model fitting and Gaussian mixture modeling, each identifying a primary Gaussian peak at 0.26 nS with a width of 0.03 nS. The same analysis was applied to the KD-RON2 conductance transients, yielding a smaller primary Gaussian peak at 0.19 nS with a width of 0.03 nS. The number of quantal units contributing to the averaged maximum conductance was not significantly different between both parasite lines. Knocking down RON2 did not eliminate the transient or the number of steps, but did change the step size significantly (27% smaller). In contrast, the step size for both the RASP2 KD and its unconditioned parent line were not significantly different from the WT step size, nor did reducing extracellular calcium change the WT step size. Thus, the elimination of RON2 both eliminated parasite invasion and altered the step size of the invasion transient in conductance.
The distinctive transient increase in host-cell membrane conductance detected after parasite attachment but preceding the morphological changes of invasion implies that, for a brief time, ions can freely move across the host-cell membrane. Ion movement (flux) is likely due to the appearance in the host-cell membrane of an aqueous pathway. Pathways for passive ion flux across membranes can be classified as either protein-lined channels (termed “protein pores”) or localized ruptures of the hydrocarbon continuity of the lipid bilayer (termed “lipidic pores”). The invasion transient proceeds along a series of intermediates in conductance: once initiated, the conductance rises in tens of milliseconds, reaching a peak before diminishing more slowly to a residual conductance that is approximately 10% of that peak (by one second after the initiation of the conductance transient). RON2 was not required for the transient increase in either membrane conductance or Ca2+ entry. Consistent with an aqueous pathway for ion flux during the conductance transient is the increasing Ca2+ flux into the host-cell cytoplasm with increasing extracellular Ca2+ concentration, which also revealed that transients require rhoptry exocytosis. However, extracellular calcium is not itself likely to be the major charge-carrying ion or required for either pore opening or closing, given that conductance transients of similar shape and magnitude were observed in an environment with twenty times lower extracellular calcium concentration. Based on these combined data, we propose that the conductance and calcium transients observed are manifestations of a rhoptry exocytosis–dependent host-cell membrane poration process (i.e., the insertion of “invasion pores”), whose purpose is to provide the pathway for rhoptry protein translocation into the host-cell cytosol. This model (see Image) is consistent with previous data showing that parasites lacking RON2 can deliver secreted rhoptry proteins into the host cytosol without complete moving junction formation.
Figure 1. Model for invasion pore formation during parasite invasion
Invasion pore formation is triggered by rhoptry exocytosis. Electrophysiological analysis supports multiple- rather than single-pore formation on the host-cell membrane. The model depicted in this figure has pore-forming proteins emerging from the mouth of the fusion pore between the apical vesicle (AV) and parasite plasma membrane to incorporate into the underlying proximal host-cell membrane. The parasite plasma membrane is orange and the host-cell plasma membrane is green. While we believe that it is likely that rhoptry proteins, perhaps after mixing with AV–resident proteins, have pore-forming activities, we cannot exclude the possibility that rhoptry proteins collaborate with resident host-cell plasma membrane proteins to form the invasion pores.
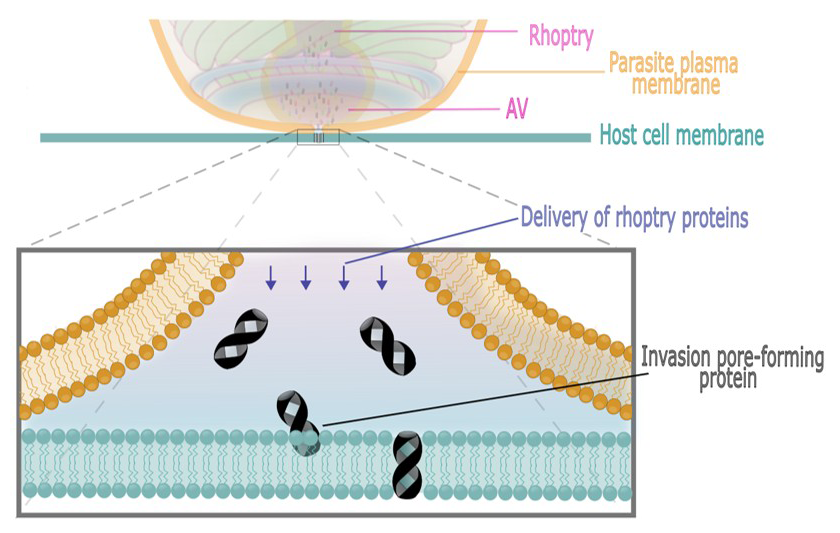
Figure 1. Model for invasion pore formation during parasite invasion
Invasion pore formation is triggered by rhoptry exocytosis. Electrophysiological analysis supports multiple- rather than single-pore formation on the host-cell membrane. The model depicted in this figure has pore-forming proteins emerging from the mouth of the fusion pore between the apical vesicle (AV) and parasite plasma membrane to incorporate into the underlying proximal host-cell membrane. The parasite plasma membrane is orange and the host-cell plasma membrane is green. While we believe that it is likely that rhoptry proteins, perhaps after mixing with AV–resident proteins, have pore-forming activities, we cannot exclude the possibility that rhoptry proteins collaborate with resident host-cell plasma membrane proteins to form the invasion pores.
The characteristics of the transient obtained from the high-resolution analysis suggest that multiple poration events occur to increase the permeability of the host-cell membrane after rhoptry exocytosis and independently of complete moving junction formation. Detailed analysis of transients revealed a fast increase in the conductance with a slower decay, each composed of multiple “steps in conductance” identified by change-point analysis. The change-point analysis supports the hypothesis that the kinetic processes for creating and removing the conductance pathways for WT and KD-RON2 are similar. The identification of abrupt changes in conductance during defined change-point mean conductances were further described using PWC analysis, where quantal conductance changes of 0.26 and 0.19 nS were identified in the WT and KD-RON2 change-point means, respectively. We propose that the observed quantal conductances arise from invasion pores whose apparent opening results in the observed rising phase (through either incorporation or stimulation by rhoptry content molecules) and whose apparent closing results in the observed falling phase (through occlusion, inactivation or gated closing) together with a flickering process (rapid opening and closing) present throughout the transient. For both parasite lines, the averaged maximum conductance is modeled as the action of 13–16 quantal units acting additively. Alternative explanations for a quantal step size are worth exploring. Multiple steps in conductance are not unusual for proteinaceous pores having subconductance states, such as the voltage-dependent anion channel (VDAC) [Zimmerberg J, Parsegian VA, Nature 1986;323:36]. The largest component in the distribution of conductance step sizes for VDAC is the main open-closed transition (also about 0.25 nS in salt solutions). A single proteinaceous pore model with 13–16 identical subconductance units (size, duration, and open and closing properties) is unlikely, barring an unusual sequential conductance state transition pattern that mimics the exponential rising and falling time distributions observed using the change-point analysis. However, our analysis does not rule out multiple proteinaceous pores with lower numbers of subconductance states whose individual stochastic behavior (subconductance states) and combined behavior (activation) result in the macroscopically observed conductance changes. But the simplest explanation of the data is near-synchronous creation of multiple pores by material secreted from the rhoptries into the host-cell plasma membrane (see Image). The function of these pores is unknown, but their similar size to protein translocons (protein complexes that move polypeptides across the ER), and their appearance in the sequence of invasion (after rhoptry exocytosis and before moving junction formation) support a hypothesis that the invasion pores create a pathway for delivery of secreted rhoptry proteins into the host-cell cytoplasm.
The physico-chemical mechanism by which a small domain of viral spike proteins allows the penetration step of viral infection
We studied the isolated fusion peptide (FP) of influenza virus, both computationally and experimentally. Our recent discovery of the aggregation of FP was investigated in more detail through molecular dynamics simulations in four different membrane compositions to examine lipid-mediated clustering and poration free energy. We confirmed the formation of highly stable antiparallel FP dimers and found them to be primarily stabilized by peptide-peptide interactions and largely insensitive to membrane composition. However, lipid sorting under the FP dimer was strongly dependent on the lipids' spontaneous curvature, with a positive spontaneous curvature-generating lipid depleted, and negative spontaneous curvature-generating lipids enriched under the dimer. In simulations of ten FP dimers, the cholesterol-containing membrane promoted higher-order clustering, seen as formation of tightly bound tetramers and linear arrangements of dimers that were not observed in the other membrane compositions. These tightly bound tetramers were associated with reduced poration efficiency compared with more loosely associated configurations, consistent with earlier experimental work showing that cholesterol partially inhibits poration by FP. These findings demonstrate how lipids with different curvature-generating propensities modulate peptide aggregation and pore formation, and may have broader implications for other enveloped viruses, such as SARS-CoV-2, where similar FP clustering has been observed. Complementing these computational studies, experiments involving labeled FP interacting with a membrane model system (supported lipid bilayers) indicate lipid compositional differences in the surface distribution of hemagglutinin in time. Cholesterol:POPC (POPC is a phospholipid) membranes display a heterogeneous distribution of FP that differs from that observed in pure POPC membranes. These differences recapitulate our previously published observations indicating differences between FP interacting with POPC vesicles and cholesterol:POPC vesicles.
Publications
- Activation of the receptor KIT induces the secretion of exosome-like small extracellular vesicles. J Extracell Biol 2024 3:e139
- Conserved sequence features in intracellular domains of viral spike proteins. Virology 2024 599:110198
- Perforation of the host cell plasma membrane during Toxoplasma invasion requires rhoptry exocytosis. EMBO Rep 2025 26:5027-5047
- The invasion pore induced by Toxoplasma gondii. EMBO Rep 2025 26:5009-5026
- Defining the EM-signature of successful cell-transfection. bioRxiv 2024 doi: 10.1101/2024.03.07.583927:preprint
Collaborators
- Vadim Frolov, PhD, Universidad del País Vasco, Bilbao, Spain
- Samuel T. Hess, PhD, University of Maine, Orono, ME
- Maryse Lebrun, PhD, Université de Montpellier, Montpellier, France.
- Sebastian Lourido, PhD, Massachusetts Institute of Technology, Cambridge, MA
- Sukbir Kaur, PhD, Laboratory of Pathology, Center for Cancer Research, NCI, Bethesda, MD
- Frances Male, PhD, University of Vermont Larner College of Medicine, Burlington, VT
- Doreen Matthies, PhD, Unit on Structural Biology, NICHD, Bethesda, MD
- Dean D. Metcalfe, MD, MS, Laboratory of Allergic Diseases, NIAID, Bethesda, MD
- Vinh-Nhan Ngo, MS, University of Maine, Orono, ME
- Richard Pastor, PhD, Laboratory of Membrane Biophysics, NHLBI, Bethesda, MD
- Annika Pfeiffer Daniels, PhD, Laboratory of Allergic Diseases, NIAID, Bethesda, MD
- Ana Olivera, PhD, Mast Cell Biology Section, NIAID, Bethesda, MD
- Prakash Raut, PhD, University of Maine, Orono, ME
- Saima M. Sidik, PhD, Whitehead Institute, Cambridge, MA
- Alexander J. Sodt, PhD, Unit on Membrane Chemical Physics, NICHD, Bethesda, MD
- Dylan Valleau, PhD, Whitehead Institute, Cambridge, MA
- Gary E. Ward, PhD, University of Vermont Larner College of Medicine, Burlington, VT
- Jack Yanovski, MD, PhD, Section on Growth and Obesity, NICHD, Bethesda, MD
Contact
For more information, email Joshua.Zimmerberg@nih.gov or visit https://irp.nih.gov/pi/joshua-zimmerberg.