



You are here: Home > Section on Metabolic Regulation
Signal Transduction in Synaptic Transmission and Plasticity

- Kuo-Ping Huang, PhD, Head, Section on Metabolic Regulation
- Freesia L Huang, PhD, Staff Scientist
- Guoxiang Liu, PhD, Visiting Fellow
We investigate the signal transduction mechanisms involved in synaptic transmission and plasticity. Studies of these neural processes are essential to understanding the complex problems related to cognition and behavioral abnormalities. Our current focus is to study genetically modified mice with deletion of the gene encoding neurogranin (Ng), which is specifically expressed in the brain. Ng is a neural-specific protein, which is normally expressed at a high level in subsets of neurons in the forebrain. This protein has been implicated in the regulation of Ca2+/calmodulin (CaM)-dependent cellular processes important for the enhancement of synaptic transmission and plasticity. In humans, mutation of Ng has been linked to behavioral abnormalities and cognitive deficits. Ng levels in the neuronal soma and dendrites are very high, and the protein sequesters apoCaM at basal physiological Ca2+. Upon synaptic stimulation, the influxed Ca2+ displaces Ng from the Ng/apoCaM complex to form Ca2+/CaM and free Ng. The buffering of CaM by Ng serves as a mechanism to regulate neuronal free Ca2+ and Ca2+/CaM concentrations. The aim of this project is to define the regulatory functions of Ng in neuronal signaling and to design therapeutic approaches to treat cognitive deficits and behavioral disturbances in children suffering from a mutation in Ng.
Ca2+-sensitive translocation of calmodulin and neurogranin in the hippocampal CA 1 neurons
Ng is a low-molecular-weight (7.5 kDa) neuronal protein expressed at high levels in the hippocampus, neocortex, and amygdala and has been shown to play an important role in the enhancement of synaptic plasticity and learning and memory. In vitro, Ng binds to apo-CaM, and the interaction is weakened by increasing Ca2+ concentration, Ng's phosphorylation by PKC, and oxidation by nitric oxide and other oxidants. The phosphorylated Ng can stimulate G protein–coupled phosphoinositide second messengers to trigger the mobilization of Ca2+ from intracellular stores. Based on yeast two-hybrid screening, CaM is believed to be an Ng binding partner in vivo. It has been suggested that Ng modulates synaptic responses by buffering and sequestering CaM to regulate the level of free Ca2+ and Ca2+/CaM complexes. We found that deletion of Ng in mice causes severe deficits in learning and memory of hippocampus-dependent spatial tasks and amygdala-dependent fear responses. Acute hippocampal slices of these mutant mice also exhibited deficits in high-frequency stimulation– (HFS) induced long-term potentiation (LTP) in the CA1 region. In the absence of Ng neurotransmitter-mediated intracellular Ca2+-transients and the down-stream signaling reactions were significantly attenuated compared with wild-type mice. Among Ng+/− mice, but less so in Ng+/+ mice, a significant relationship exists between the hippocampal levels of Ng and performances in several cognitive tasks. Recently, it was reported that deletion of a copy of Ng gene from human chromosome 11q was associated with deficits in cognitive function of these patients. Association studies in schizophrenia have also linked a mutation of the Ng gene to this disease.
Fluorescence immunohistochemical staining of hippocampal slices bathed in Ca2+-containing artificial cerebral spinal fluid (ACSF) revealed that Ng and CaM were co-localized in the soma and dendrites of principle neurons. In the CA1 region, the concentrations of CaM and Ng in the soma were greater than in the dendrites. Surprisingly, majority of somatic CaM was sequestered in the nucleus. In contrast, Ng was abundantly present in the soma as well as in the dendrites. Removing Ca2+ from the bathing fluid resulted in a suppression of synaptic transmission with a concomitant redistribution of CaM and Ng from soma to dendrites. Confocal Ca2+-imaging showed that a reduction of about 15 and 40 nM [Ca2+]i were sufficient to cause half-maximum translocation of Ng and CaM, respectively, from soma to dendrites. Switching the bathing fluid back to the Ca2+-containing ACSF restored synaptic transmission and the original compartmentalization of these two proteins. The hippocampal CA1 pyramidal neurons were the more responsive to this Ca2+-sensitive translocation than their neighboring CA2 and CA3 neurons. These studies illustrated the unique features of hippocampal CA1 neurons in sequestering high concentrations of CaM and Ng in the soma and releasing them to the dendrites at reduced levels of [Ca2+]i. The Ca2+-sensitive movement of Ng and CaM between soma and dendrites highlights the unique sensitivity of CA1 pyramidal neurons to changing [Ca2+]i.
Synaptic stimulation causes mobilization of CaM from soma to dendrites.
The concentration of Ng in the hippocampus was approximately twice that of CaM and the latter protein was largely concentrated in the nucleus and much less in the dendrites. As described above, the asymmetrical distribution of CaM in hippocampal neurons was in contrast to that of Ng, which was abundantly present in the soma and dendrites. Thus, in the distal dendrites, binding of a low concentration of CaM by a relatively higher concentration of Ng renders very little CaM available for the activation of CaM-dependent enzymes. We explored the possibility that the HFS could trigger the mobilization of CaM from soma to dendrites to activate CaMKII and adenylate cyclases for the maintenance of LTP. Tetanic stimulation (a single train of 1 s, 100 Hz) of the Schaffer collateral fiber of the hippocampal slices caused an increase of CaM in the dendrites. HFS-mediated mobilization of CaM was detected surrounding the stimulating electrode within the stratum radiatum but not near a second electrode that didn’t deliver HFS. Both CaM and Ng were found to associate with the dendritic spines near the stimulating electrode. Mobilization of CaM from soma to dendrites was also observed in phorbol ester–induced synaptic facilitation in these tissues. It appears that synaptic stimulation could also trigger exit of CaM from soma (nucleus) to dendrites. These findings suggest that association of CaM and Ng at the stimulated dendritic spines may enhance synaptic efficacy by increasing the Ca2+ transients, as predicted by the “mass-action mechanism.” The association of CaM and Ng at stimulated synapses may also serve as synaptic tags for stimulated dendritic branches.
Induction of synaptic facilitation and drug treatment of the neurogranin knockout mice
Further characterization of Ng KO mice revealed that these animals exhibited additional behavioral abnormalities, including hyperactivity, inattentiveness, and impulsivity—behavioral phenotypes that likely result from disruption of Ng-regulated signaling. One of the most prominent roles of Ng is the enhancement of the NMDA-receptor mediated Ca2+-transients. Thus, we predict that stimulation of downstream signaling components post Ca2+ influx or increase in presynaptic transmitter release may rescue the deficits of Ng KO mice. In neurons, stimulation of PKCs is known to enhance transmitter release and facilitate synaptic responses. Treatment of hippocampal slices from Ng KO mice with the PKC–activating phorbol ester phorbol-12, 13-dibutylate (PDBu) caused persistent synaptic facilitation. The PDBu–mediated effects were most prominent among those tissue slices from the dorsal hippocampus, which is an area thought to be associated with cognitive functions.
In conjunction with these in vitro studies, we treated Ng KO mice with Ritalin®, a psychostimulant drug known to increase the extracellular neurotransmitters. After three weeks of daily treatment (10 mg/kg/day, i.p.), four groups of animals, including control and drug-treated wild-type and Ng KO mice, were subjected to behavioral tests. The drug-treated Ng KO mice exhibited improvement in their cognitive functions, as evidenced by a reduction of the latency time to locate the hidden platform in the water maze and an increase in the freezing time after fear conditioning. Ritalin® also appeared to reduce the hyperactivity of the Ng KO in the open field and increase the immobility time in the forced-swim chamber. The drug treatment, however, had only a marginal effect on the performance of the wild-type mice. Measurement of the HFS– (1 x 100 Hz for 1s) induced LTP in the hippocampal CA1 region in vitro showed a positive effect of the drug on the Ng KO. These results indicate that Ritalin®, a drug commonly used for the treatment of attention-deficit hyperactivity disorder (ADHD), can exert beneficial effects on the Ng KO, as it does for the human patients. These studies also suggest that Ng KO mice could serve as an animal model for ADHD and will thus be useful for the development of new treatment strategies for this illness.
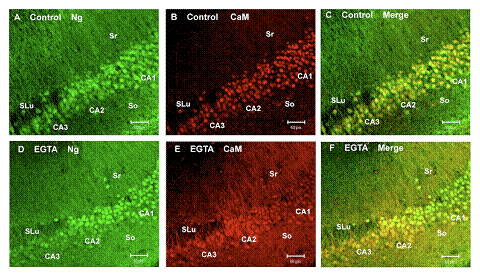
Distinctive Ca2+-sensitive translocation of Ng and CaM among the hippocampal neurons in the CA 1 versus those in the CA 2 and CA 3 regions.
The figure shows confocal images (10x) of hippocampal slices doubly stained with antibodies against CaM (red) and Ng (green) covering a population of cells in the CA1, CA2, and CA3 regions of the control (A, B, and C) and those treated with EGTA (D, E, and F) to reduce the intracellular Ca2+ by 15-40 nM. Both the CA2 and CA3 neurons appear slightly larger than those of the CA1. In the merged images of control (C), CaM co-localizes with Ng in the nucleus with a relatively low abundance in the cytoplasm and dendrites within stratum radiatum (Sr) and stratum oriens (So). In the EGTA-treated tissue (F), CaM was greatly reduced in the soma and translocated to the distal dendrites of the CA1 neurons, whereas in the CA2 and CA3 CaM remained in the soma and proximal dendrites. Mixed populations of CA1 and CA2 neurons were clearly distinguished in this EGTA-treated tissue.
Publications
- Shetty PK, Huang FL, Huang K-P. Ischemia-elicited oxidative modulation of Ca2+/calmodulin-dependent protein kinase II. J Biol Chem 2008 283:5389-5401.
Collaborators
- Alfred L. Yergey, PhD, Mass Spectrometry Core Facility, NICHD, Bethesda, MD
Contact
For more information, email kphuang@helix.nih.gov or visit http://neuroscience.nih.gov/Lab.asp?Org_ID=364