


You are here: Home > Section on Macromolecular Recognition and Assembly
Intermolecular Forces, Recognition, and Dynamics

- Donald C. Rau, PhD, Head, Section on Macromolecular Recognition and Assembly
- Nina Yu Sidorova, PhD, Staff Scientist
- Jason DeRouchey, PhD, IRTA Fellow
- Rene Royal, BA, Postbaccalaureate Fellow
- David Thomas, Summer Intern
Our laboratory focuses on elucidating the coupling of the forces, structure, and dynamics of biologically important assemblies. The next challenge in structural biology is to understand the physics of interactions between molecules in aqueous solution. The ability to take advantage of the increasing number of available protein and nucleic acid structures will depend critically on establishing a link between structures and binding energies. A fundamental and quantitative knowledge of intermolecular forces is necessary to understand the interactions among biologically important macromolecules that control cellular function and to rationally design agents that can effectively compete with those interactions associated with disease. Our results have shown that experimentally measured forces at close spacings are very similar for a wide variety of systems, but very different from those predicted by current, conventionally accepted theories. We have interpreted the observed forces as indicating the dominating contribution from water-structuring energetics. The observation that interacting macromolecules will tenaciously retain their hydration waters unless the surfaces are complementary has profound implications for recognition reactions. To investigate the role of water in binding, we measure and correlate changes in binding energies and hydration accompanying recognition reactions of biologically important macromolecules, particularly of sequence-specific DNA-protein complexes. We observe a strong correlation between retained water and binding energy; stronger binding means less water retained at the DNA-protein interface.
Direct force measurements
The ability to measure directly forces between biopolymers in macroscopic condensed arrays has greatly changed our understanding of how molecules interact at close spacings—the last 1-1.5 nanometers of separation. The universality of the force characteristics observed for a wide variety of macromolecules (Figure 1), charged or uncharged, including DNA, proteins, lipid bilayers, and carbohydrates, and for the interaction of small solutes and salts with macromolecules has led us to conclude that the energy associated with changes in structuring water between surfaces dominates intermolecular forces. We directly measure forces between biological macromolecules in macroscopic condensed arrays using osmotic stress and x-ray scattering.
DNA-DNA attractions
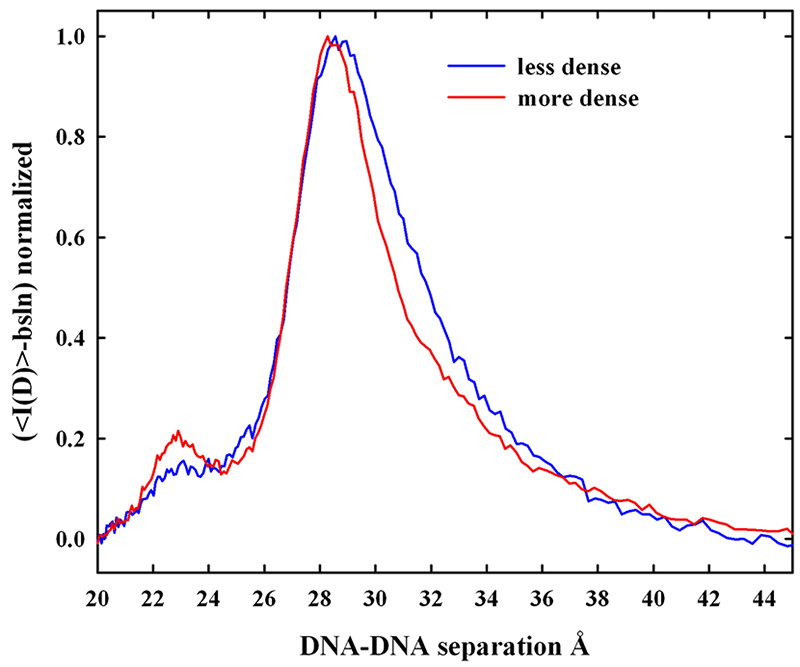
Figure 1: A common repulsive force for varied biological systems
A common exponentially varying dependence of the distance between molecules and the applied pressure is seen for very different biosystems—charged (DDP - a charged lipid bilayer, DNA, and i-carrageenan), zwitterionic (egg PC bilayers), and uncharged (schizophyllan and hydroxypropyl cellulose - HPC). The exponential decay length is about 0.4 - 0.5 nm. The excess pressures applied by 1 M methylpentanediol on DNA and by 1 M sorbitol on HPC are also shown to illustrate that the interaction of small molecules—charged or uncharged—with macromolecules—charged or uncharged—also follows the same exponential dependence. The force amplitudes of the exponentials vary by over an order of magnitude and are presumed to depend on the strength of water structuring on the macromolecular surfaces. (click image to enlarge)
Dense packaging of DNA in viruses, bacteria, eukaryotic nuclei, and sperm heads is necessary for proper cellular functioning. DNA assembly caused by multivalent ions is a critical testing ground for understanding not only in vivo compaction of DNA but also the physics of interactions between charged molecules. If multivalent ions are present at a sufficiently high concentration, DNA will spontaneously assemble into an ordered array. The helices do not collapse to touching but are rather separated by 0.5-1.5 nm of solvent depending on the nature of the condensing ion. Attractive and repulsive forces balance at the equilibrium spacing. We described the attractive and repulsive forces for several commonly used condensing agents and for a series of oligo-arginine peptides. The attraction is characterized by an exponential force with a 0.5 nm decay length. Attraction results from the direct interaction of correlated and complementary surface hydration structures. From the experimental results for a set of homologous oligo-arginine peptides, we determined that the magnitude of the attractive force increases with the number of charges, consistent with a constant loss in entropy from correlating a single molecule regardless of charge, but a gain in interaction energy with the number of charges. The residual repulsion is also characterized by an exponential force, but with one-half the decay length (0.25 nm) of the attractive force. The interaction is best described as the hydration equivalent of the image charge repulsive force in electrostatics. The hydration atmosphere extending from a solvated surface stabilizes water structuring at the surface. Disruption of the hydration atmosphere of one surface by another surface will lower hydration energies regardless of the water structuring on the other surface.
We have extended this work and are now investigating condensed arrays of protamine-assembled DNA. Protamines are small (30-60 aa), arginine-rich (55-65%) peptides used to package DNA in vertebrate sperm heads. Protamine-DNA forces resemble the forces seen for all other condensing ions that we measured, spanning the range from simple metal ions as Mn2+ and Co3+, to the biogenic amines spermidine3+ and spermine4+, finally to the oligo-arginine peptide series (2+ to 6+). There is no additional contribution from the protein character of protamines. The magnitude of the repulsive force is significantly larger than expected from the oligo-arginine peptide measurements, suggesting that the neutral amino acids present in protamine result in an increased residual repulsion. We confirmed this hypothesis by measuring DNA force curves with synthetic hexa-arginines that incorporate increasing numbers of neutral amino acids. The 0.25 m decay-length repulsive-force amplitude increases linearly with the number of inserted alanines (0-4). The character of the neutral amino acid from hydrophilic serines to hydrophobic isoleucines does make a difference to the overall force magnitude.
We also measured DNA assembly forces in salmon sperm nuclei. The in vivo sperm and in vitro reconstituted salmon protamine-DNA forces are virtually identical. These force measurements thus have direct biological application. Mammalian protamines are more complicated than piscine; two protamine species are typically present, and disulphide bridges form between protamines, presumably to further condense DNA. This can be easily verified by measuring DNA-DNA spacing with and without added disulphide reducing agents. We will optimize our experimental protocols with bull sperm before investigating human sperm.
We can use our knowledge and unique experimental tools to investigate the connection between DNA packaging in sperm heads, DNA damage, and male infertility. There are several reports in the literature correlating human male infertility with protamine abnormalities and misprocessing. Cumulative DNA damage from reactive oxygen species is a major contributor to male infertility given the absence of DNA repair systems from sperm. We hypothesize that incorrect assembly of DNA by insufficient or modified protamines will result in less tightly packaged DNA in sperm (a larger than normal distance between helices) and, consequently, a greater accessibility of oxidizing free radicals to DNA. We can test this hypothesis by direct measurements of the spacing between helices in sperm of normal and infertile males and of accumulated DNA damage.
The replacement of histones by protamines in spermatogenesis occurs in several steps. Before binding to DNA, several of the protamine serines are first phosphorylated. Then phosphorylated protamine-DNA complexes are dephosphorylated and the DNA is fully packaged. We examined the effect of phosphorylation of synthetic protamine-like peptides on DNA-DNA forces. As expected from previous measurements, R3SR3 is only slightly different from R6. The phosphorylated peptide, R3pSR3, however, profoundly weakens attraction, increasing the distance between DNA surfaces from 0.85 nm to 1.35 nm, even in the absence of any competing salt that would be found in the cell. This level of phosphorylation mimics that found in naturally occurring protamines. The greatly weakened attraction suggests that the function of protamine phosphorylation is to prevent tight packaging of DNA and to allow phosphatase access. An orderly dephosphorylation of these modified protamines would consequently allow an orderly condensation of DNA. Incomplete dephosphorylation and, therefore, less tightly assembled DNA have been linked to infertility.
Hydration changes linked to sequence-specific DNA-protein recognition reactions
Our ultimate goal is to apply the lessons from direct force measurements to the recognition reactions that control cellular processes. We focused on differences in water sequestered by complexes of sequence-specific DNA-binding proteins bound to different DNA sequences, with particular emphasis on correlating binding energy and water incorporated and on measuring the energy necessary to remove hydrating water from complexes. We determine differences in sequestered water between complexes through the effect of osmotic pressure on binding constants or dissociation rates. Differences in water between specific and nonspecific complexes connect thermodynamics and structure. The emphasis on water allows us to approach protein-DNA protein binding from a direction that differs from standard practice. Indeed, we find that sequestered water correlates quite well with binding energy and constitutes the key difference between specific, non-cognate, and nonspecific complexes.
EcoRV binding to specific and non-specific DNA sequences
We focused on DNA complexes of restriction endonucleases in particular. Beyond their unparalleled importance as tools for the analysis and manipulation of DNA, restriction enzymes have proven a remarkable model system for uncovering and analyzing numerous aspects of protein-nucleic acid interactions. The proteins combine high binding strength and extraordinary sequence specificity. We take advantage of the unique experimental tools we have developed to investigate the coupling of structure, thermodynamics, and function of these complexes from a fresh perspective.
We are currently investigating DNA complexes of the type II restriction enzyme EcoRV. Typically this class of restriction endonucleases can distinguish between specific recognition and nonspecific DNA sequences quite efficiently. Results in the literature, however, indicate that EcoRV has an unusually low sequence stringency, with less than a 10-fold difference between EcoRV binding constants to specific and nonspecific sequences at pH 7.5. Solved X-ray crystal structures for specific and non-cognate DNA-EcoRV complexes differ noticeably. The interface of the specific complex is essentially anhydrous with many direct DNA-protein interactions and is very different from the non-cognate complex that has a large water-filled gap at the protein-DNA interface. The difference in structures is comparable to that seen for BamHI complexes. The differences would suggest significant differences in hydration and in specific-nonspecific binding free energies between two EcoRV complexes, as we measured for BamHI complexes. We applied a self-cleavage assay that we had developed to measure EcoRV-DNA binding in solution. The technique monitors only enzymatically competent complexes of the endonuclease and, while providing the same level of sensitivity, is not subject to the limitations of gel mobility shift assay.
Unexpectedly, we found that EcoRV has unusual kinetics for specific complex formation. A significant fraction of the total enzyme is in a conformation that forms enzymatically competent complexes unusually slowly (hours) at pH 7.5. The slow rate of complex formation can explain the lack of specificity reported by others. When we measured the ratio of specific and nonspecific binding constants that provide the abnormally long incubation times necessary to achieve equilibrium, we found that the binding of EcoRV to the recognition sequence is about 1000-fold stronger than to a nonspecific sequence at pH 7.5. This is more than an order of magnitude larger than has been typically observed. The relative specific-nonspecific binding constant Knsp-sp increases strongly with decreasing pH and with increasing neutral osmolyte concentration. The osmotic pressure dependence of the relative binding constant, however, is only weakly sensitive to pH, indicating that the structures of the specific and nonspecific complexes likely change minimally with pH. The difference in the number of water molecules sequestered by the specific and nonspecific complexes is consistent with the size of the water-filled gap at the protein-DNA interface of the nonspecific complex. There is also a change in the solute-exposed surface area of the protein that is particularly apparent with the osmolyte triethylene glycol.
As we have emphasized in the past, the large osmotic dependence of Knsp-sp means that measurement of protein-DNA specificity in dilute solution cannot be directly applied to binding in the crowded environment of the cell. In addition to divalent ions, water activity and pH are two key parameters that strongly modulate binding specificity of the EcoRV.
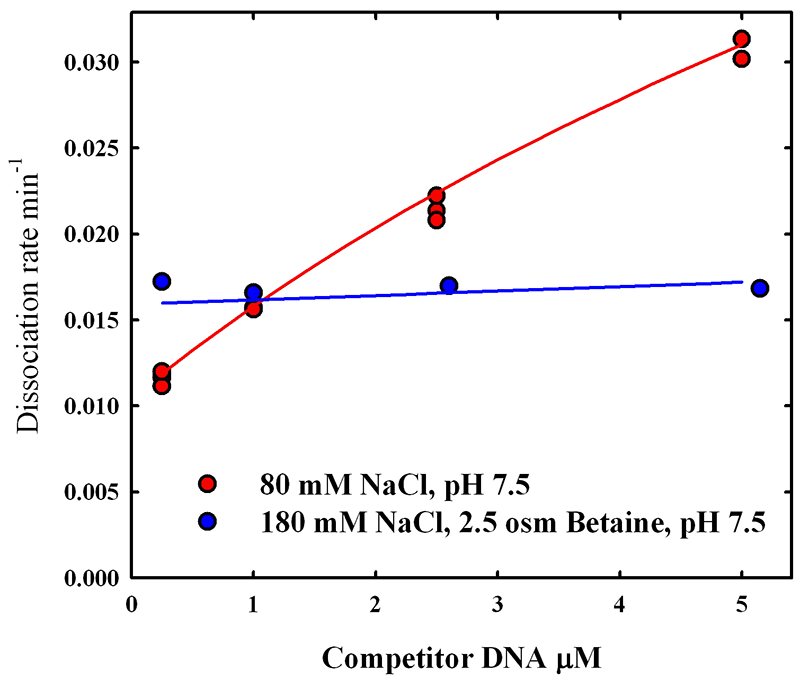
Figure 2. Allosteric conformers of EcoRV
The closed-arm structure is that seen in the x-ray crystallographic structure of the free protein. The open-arm conformation was taken from the structure of the nonspecific complex. (click image to enlarge)
The observation of at least two components in the association kinetics indicates that EcoRV is an allosteric protein with at least two conformers. Allosterism is now recognized as important concept for DNA-protein complexes, offering an additional level of control over binding and activity. The recognition specificity or activity of DNA-binding proteins can be modulated by ligands or proteins that bind to one allosteric conformation in preference to others. We are continuing our investigation into the EcoRV structures responsible for the different kinetic classes of association. Our working hypothesis is that the free protein exists both in a conformation with open DNA-enveloping arms that is able to bind to DNA and in a conformation with closed arms that is sterically unable to bind to DNA (Figure 2). The closed-arm conformation is the structure seen in the crystal structure of free EcoRV. We are currently determining the dependence of the equilibrium distribution of the two protein structures and their exchange kinetics on pH, salt concentration, osmotic pressure, and divalent cation concentration.
Stabilizing labile DNA-protein complexes in polyacrylamide gel using osmolytes
In addition to its use as a thermodynamic variable, osmotic stress has several practical applications. Osmotic pressure can greatly stabilize complexes, slowing dissociation rates by orders of magnitude. We used osmotic pressure to stabilize labile complexes in polyacrylamide gels. The electrophoretic mobility assay (EMSA) is widely used to monitor the formation of DNA-protein or RNA-protein complexes. The mobility of complexes and free DNA typically differ in polyacrylamide gel electrophoresis. Many nonspecific DNA-protein complexes, however, are sufficiently weak that they dissociate while migrating, giving smeared bands that are difficult to quantitate. We find that adding osmolytes, in particular triethylene glycol, directly to the gel can dramatically stabilize noncognate and nonspecific complexes of the restriction nuclease EcoRI. It is straightforward to extend this approach to other techniques, such as gel chromatography and capillary electrophoresis, for the separation of complex and free components.
Publications
- Todd B, Stanley C, Sidorova NY, Rau DC. Hydration forces: water and biomolecules. In: Wiley Encyclopedia of Chemical Biology 2009;1200-1225.
- Rau DC, Sidorova NY. Diffusion of the restriction nuclease EcoRI along DNA. J Mol Biol. 2010;395:408-416.
- Sidorova NY, Hung S, Rau DC. Stabilizing labile DNA-protein complexes in polyacrylamide gels. Electrophoresis. 2010;31:648-653.
- DeRouchey J, Parsegian VA, Rau DC. Cation charge dependence of the forces driving DNA assembly. Biophys J. 2010;99:2608-2615.
Collaborators
- Jason DeRouchey, PhD, Department of Chemistry, University of Kentucky, Lexington, KY
- William Gelbart, PhD, University of California Los Angeles, Los Angeles, CA
- Michael Mossing, PhD, Department of Chemistry and Biochemistry, University of Mississippi, Oxford, MS
- V. Adrian Parsegian, PhD, Department of Physics, University of Massachusetts, Amherst, MA
- Xiangyun Qiu, PhD, Department of Physics, George Washington University, Washington, DC
- T.J. Thomas, PhD, University of Medicine and Dentistry of New Jersey–Robert Wood Johnson Medical School, Piscataway, NJ
- William Whittier, DVM, Department of Large Animal Clinical Sciences, Virginia-Maryland Regional College of Veterinary Medicine, Blacksburg, VA
Contact
For more information, email raud@mail.nih.gov.