

You are here: Home > Section on Intracellular Protein Trafficking
Protein Sorting in the Endosomal-Lysosomal System

- Juan S. Bonifacino, PhD, Head, Section on Intracellular Protein Trafficking
- Rafael Mattera, PhD, Staff Scientist
- Xiaolin Zhu, RN, Technician
- Xuefeng Ren, PhD, Research Fellow
- Loreto Cuitiño, PhD, Visiting Fellow
- Ginny Farias, PhD, Visiting Fellow
- Xiaoli Guo, PhD, Visiting Fellow
- Shweta Jain, PhD, Visiting Fellow
- Sang-Yoon Park, PhD, Visiting Fellow
- Jing Pu, PhD, Visiting Fellow
- Christina A. Schindler, PhD, Visiting Fellow
- Cecilia Bahamon, BS, Postbaccalaureate Student
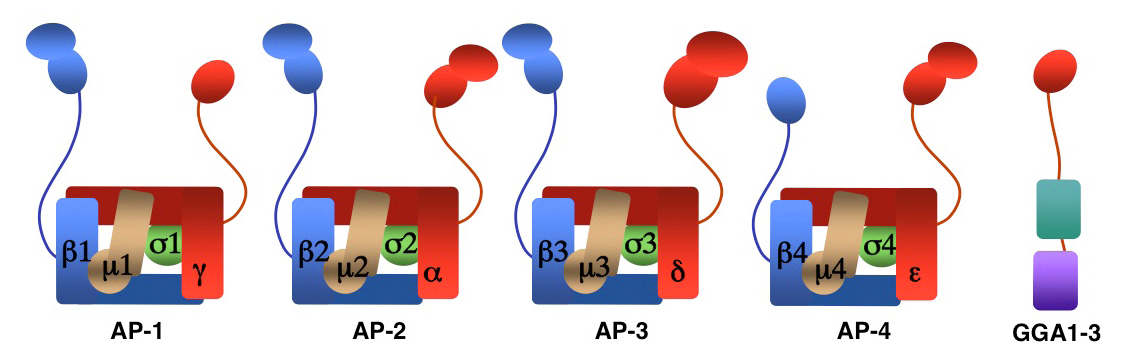
We investigate the molecular mechanisms by which transmembrane proteins are sorted to different compartments of the endomembrane system such as endosomes, lysosomes, and a group of cell-type–specific organelles known as lysosome-related organelles (e.g., melanosomes and platelet dense bodies). Sorting is mediated by recognition of signals present in the cytosolic domains of the transmembrane proteins by adaptor proteins that are components of membrane coats (e.g., clathrin coats). Among these adaptor proteins are the heterotetrameric AP-1, AP-2, AP-3, and AP-4 complexes, the monomeric GGA proteins (Figure 1), and the heteropentameric retromer complex. Proper sorting requires the function of additional components of the trafficking machinery that mediate vesicle tethering and fusion. Current work in our laboratory aims to elucidate the structure, regulation, and physiological roles of coat proteins and vesicle-tethering factors, and to investigate human diseases that result from genetic defects in these proteins (e.g., Hermansky-Pudlak syndrome; neurodegenerative and neurodevelopmental disorders).
Role of adaptor protein complexes in the sorting of the beta-site amyloid precursor protein–cleaving enzyme 1 and the oculocutaneous albinism type 2 protein
Click image to enlarge.
Figure 1. Structure of AP complexes and GGAs
AP-1, AP-2, and AP-3 are clathrin-associated adaptor protein complexes that recognize two types of sorting signal, which are referred to as tyrosine-based and dileucine-based sorting signals. Previous studies from our laboratory showed that tyrosine-based signals bind to the μ1, μ2, and μ3 subunits, whereas dileucine-based signals bind to a combination (i.e., a hemicomplex) of two subunits, γ-σ1, α-σ2 or δ-σ3, from the corresponding AP complexes. This past year, we demonstrated that a dileucine-based sorting signal in the cytosolic tail of the beta-site amyloid precursor protein (APP)–cleaving enzyme 1 (BACE1) interacts with AP-2 to mediate rapid endocytosis and lysosomal targeting of the protein. BACE1 endocytosis, however, is dispensable for APP cleavage, supporting the notion that the pathogenic amyloid-beta peptide is generated in the late secretory pathway rather than in endosomes (1). In addition, in collaboration with the group of Michael Marks, we showed that recognition of another dileucine-based sorting signal in the cytosolic tail of the oculocutaneous albinism type 2 (OCA2) protein by AP-1 and AP-3 mediates OCA2 sorting to melanosomes.
An AP-1/clathrin pathway for the sorting of transmembrane receptors to the somatodendritic domain of hippocampal neurons
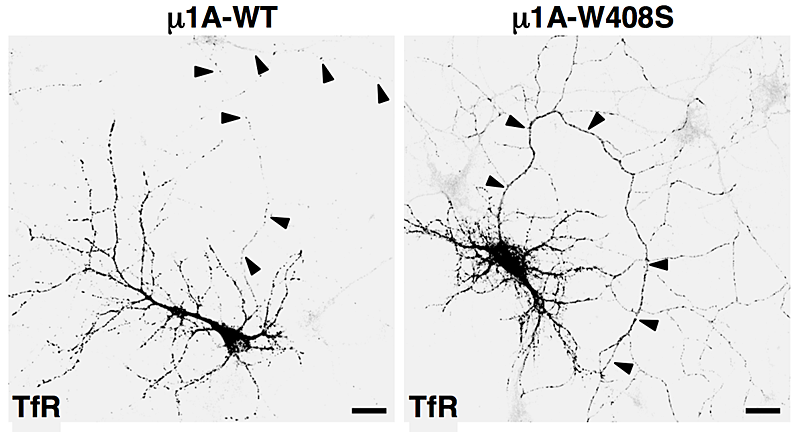
Click image to enlarge.
Figure 2. Overexpression of dominant-negative mutant of AP-1 subunit (μ1A-W408S) causes missorting of transferrin receptor (TfR) to the axon (arrowheads).
A major effort of our laboratory this past year was to examine the role of signal-adaptor interactions in polarized sorting in neurons (2). Neurons are polarized into dendrites, soma, and axons. The plasma membrane of each of these domains possesses a distinct set of transmembrane proteins, including receptors, channels, transporters, and adhesion molecules. We hypothesized that sorting to these domains could be mediated by interaction of sorting signals with AP complexes. Our studies showed that the cytosolic tails of various transmembrane receptors, including the transferrin receptor (TfR), the Coxsackie virus and adenovirus receptor (CAR), and the glutamate receptor proteins mGluR1, NR2A, and NR2B, all have information leading to their sorting to the somatodendritic domain of hippocampal neurons. In the case of TfR and CAR, this information exists in the form of tyrosine-based sorting signals. Protein interaction analyses showed that the tails of these receptors bind to the μ1A subunit of AP-1. Dominant-negative interference and RNAi approaches demonstrated that interaction of cytosolic tails with AP-1 was responsible for somatodendritic sorting. For example, overexpression of a dominant-negative mutant of the μ1A subunit of AP-1 (μ1A-W408S), but not of the wild-type protein (μ1A-WT), resulted in the appearance of transferrin receptor (TfR) in the axon (Figure 2). Sorting involved exclusion of the receptor proteins from transport carriers destined for the axonal domain at the level of the soma. Interference with AP-1–dependent somatodendritic sorting caused defective maturation of dendritic spines and reduced the number of synapses. Recently, mutations in the sigma1A and sigma1B subunits of AP-1 were shown to be the cause of two neurodevelopmental disorders known as MEDNIK syndrome and Fried syndrome, respectively. Our findings suggest that these disorders may arise from failure to sort certain cargoes to the somatodendritic domain of specific neuronal populations.
Role of the AP-1 adaptor in protein sorting to the basolateral domain of polarized epithelial cells
Epithelial cells are also polarized into basolateral and apical domains. We collaborated with the group of Enrique Rodriguez-Boulan to demonstrate that interaction of tyrosine-based sorting signals with the μ1A and μ1B subunits of AP-1 mediates sorting of transmembrane proteins such as TfR and CAR to the basolateral domain of polarized epithelial cells (3, 4). Taken together, studies with neurons and epithelial cells indicate that the AP-1 complex is a global regulator of cell polarity.
Mechanisms of CD4 downregulation by the Vpu protein of HIV-1
Our laboratory also investigates the mechanisms by which the Vpu protein encoded by the human immunodeficiency virus-1 (HIV-1) genome downregulates the CD4 co-receptor from the surface of infected cells. Specifically, we aim to identify the host-cell factors required for downregulation and to explain the molecular mechanisms involved. Primate immunodeficiency viruses target helper T cells and macrophages/monocytes through binding of the viral envelope glycoprotein to a combination of CD4 and a chemokine receptor (CCR4 or CXCR5) on the surface of the host cells. Strikingly, infection results in rapid and sustained downregulation of CD4 and, to a lesser extent, of the chemokine receptors. Downregulation of these viral co-receptors prevents superinfection, promotes virion release, and interferes with the immune response, leading to the establishment of a robust infection. CD4 downregulation is so important to the life cycle of HIV-1 that two accessory proteins, Nef and Vpu, encoded by the viral genome, are devoted to this task. Indeed, Nef and Vpu are critical for the progression from infection to AIDS, a fact that is best illustrated by the existence of long-term non-progressors who are infected with HIV-1 strains bearing inactivating mutations in the genes encoding these proteins. Therefore, pharmacologic or biologic perturbation of Nef and/or Vpu has the potential to prevent the pathogenic effects of HIV-1. To date, however, this potential has not been realized, mainly because Nef and Vpu have no enzymatic activity and their mechanisms of action are insufficiently understood.
This past year, we discovered that the transmembrane domains (TMD) of both Vpu and CD4 are required for Vpu-induced CD4 downregulation (5). We found that Trp22 in the Vpu TMD and Gly415 in the CD4 TMD are critical for Vpu-induced targeting of CD4 to endoplasmic reticulum (ER)–associated degradation (ERAD). Vpu Trp22 promotes CD4 polyubiquitination and subsequent recruitment of the VCP-UFD1L-NPL4 dislocase complex. In the presence of a Vpu Trp22 mutant, CD4 remains integrally associated with the ER membrane, suggesting that dislocation from the ER into the cytosol is impaired. CD4 Gly415, on the other hand, contributes to CD4–Vpu interactions. We also identified two residues, Val20 and Ser23, in the Vpu TMD that mediate retention of CD4 in the ER. Our findings highlight the exploitation of several TMD–mediated mechanisms by HIV-1 Vpu in order to downregulate CD4 and thus promote viral pathogenesis.
Additional Funding
- Intramural AIDS Targeted Antiviral Program (IATAP)
Publications
- Prabhu Y, Burgos PV, Schindler C, Farías G, Magadán JG, Bonifacino JS. Adaptor protein-2-mediated endocytosis of the β-secretase BACE1 is dispensable for amyloid precursor protein processing. Mol Biol Cell 2012;23:2339-2351.
- Farías GG, Cuitino L, Guo X, Ren X, Jarnik M, Mattera R, Bonifacino JS. Signal-mediated, AP-1/clathrin-dependent sorting of transmembrane receptors to the somatodendritic domain of hippocampal neurons. Neuron 2012;75:810-823.
- Gravotta D, Carvajal-Gonzalez JM, Mattera R, Deborde S, Banfelder J, Bonifacino JS, Rodriguez-Boulan E. The clathrin adaptor AP-1A mediates basolateral polarity. Dev Cell 2012;22:811-823.
- Carvajal-Gonzalez JM, Gravotta D, Mattera R, Diaz F, Perez-Bay A, Roman AC, Schreiner R, Thuenauer R, Bonifacino JS, Rodriguez-Boulan E. Basolateral sorting of CAR through interaction of a canonical YXXΦ motif with the clathrin adaptors AP-1A and AP-1B. Proc Natl Acad Sci USA 2012;109:3820-3825.
- Magadán JG, Bonifacino JS. Transmembrane domain determinants of CD4 downregulation by HIV-1 Vpu. J Virol 2012;86:757-772.
Collaborators
- Aitor Hierro, PhD, CIC-bioGUNE, Bilbao, Spain
- James Hurley, PhD, Laboratory of Molecular Biology, NIDDK, Bethesda, MD
- Michael S. Marks, PhD, University of Pennsylvania School of Medicine, Philadelphia, PA
- Enrique Rodríguez-Boulan, MD, Weill Cornell Medical College, New York, NY
Contact
For further information, contact juan@helix.nih.gov or visit www.nichd.nih.gov/research/atNICHD/Investigators/bonifacino.