

You are here: Home > Unit on Microbial Pathogenesis
Host Cell Modulation by the Intracellular Pathogen Legionella pneumophila

- Matthias Machner, PhD, Head, Unit on Microbial Pathogenesis
- Kimberly Decker, PhD, Postdoctoral Fellow
- Andrew Gaspar, PhD, Postdoctoral Fellow
- M. Ramona Neunuebel, PhD, Postdoctoral Fellow
- Timothy Evans, BSc, MSc, Postbaccalaureate Student
Many Gram-negative bacteria use type IV secretion systems (T4SSs) to deliver bacterial effector proteins into host cells, where they modulate signaling events to create an environment supportive of bacterial growth. One of those microbes is Legionella pneumophila, the causative agent of a life-threatening pneumonia called Legionnaires' disease. Upon phagocytosis by alveolar macrophages, L. pneumophila delivers a large number of bacterial proteins, or effectors, into the host-cell cytosol through the Dot/Icm T4SS (Figure 1). Some effectors help L. pneumophila redirect proteins and membrane material of the infected cell to its Legionella-containing vacuole (LCV), thereby establishing a protective compartment resembling host-cell endoplasmic reticulum (ER). L. pneumophila mutants with a non-functional T4SS are avirulent, underscoring the importance of the translocated effector proteins for L. pneumophila infection. The main focus of our laboratory is to identify and characterize host-pathogen interactions during L. pneumophila infection and to determine their importance for bacterial virulence.
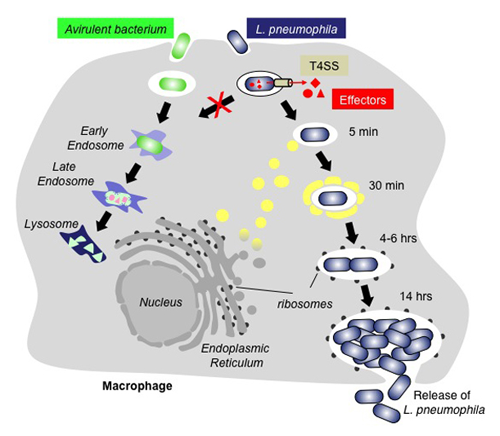
Figure 1. The infection cycle of L. pneumophila
Intracellular trafficking of Legionella pneumophila (blue) or an avirulent bacterium (green). Upon uptake by a macrophage cell, the phagosome containing an avirulent bacterium traffics along the endosomal route and gradually matures into a lysosome, which kills and degrades the bacterium. In contrast, L. pneumophila can escape the default endo-lysosomal pathway with the help of its effector proteins (red) that are infected into the host cell through the Dot/Icm type IV secretion system (T4SS; beige). Proteins and transport vesicles (yellow spheres) are recruited to the Legionella-containing vacuole (LCV), which is slowly transformed into a ribosome-studded compartment that mimics host-cell rough endoplasmic reticulum (ER) and supports replication of L. pneumophila. Eventually, the host cell is lyzed, and L. pneumophila bacteria are released to infect other macrophages.
Exploitation of the host-cell GTPase Rab1 by L. pneumophila effector proteins
Click image to enlarge.
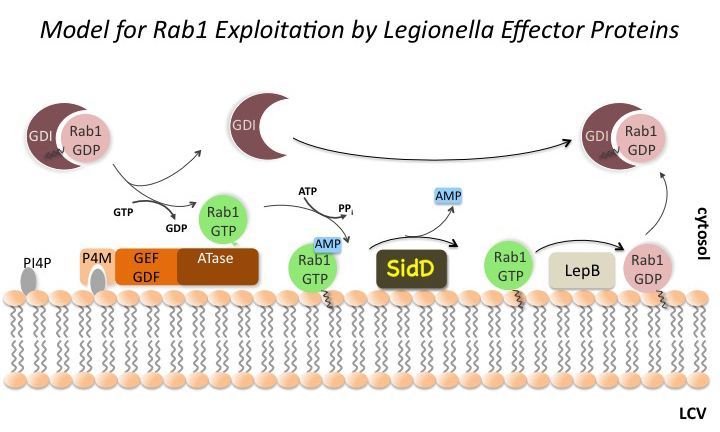
Figure 2. The Rab1 cycle on LCV membranes
One of the trafficking routes from which vesicles are intercepted by L. pneumophila is the early secretory pathway between the ER and the Golgi compartment. Rab1, a key regulator of this pathway, transiently localizes to the LCV (Figure 2), suggesting that L. pneumophila utilizes Rab1 in order to redirect secretory vesicle traffic to its LCV. We recently discovered that the effector protein SidM is critical for Rab1 exploitation. SidM combines the activity of a GDI displacement factor (GDF) with a guanine nucleotide exchange factor (GEF). SidM mediates dissociation of Rab1 from its chaperone GDP–dissociation inhibitor (GDI) and Rab1 activation by catalyzing exchange of GDP for GTP in Rab1. Bacterial mutants lacking SidM are defective for Rab1 recruitment, indicating that SidM is essential to exploit the pool of GDI–bound Rab1 during infection. SidM is the first known example of a protein shown to combine GDI displacement and nucleotide exchange activity. It remains to be seen whether other prokaryotic and eukaryotic proteins exist that share this remarkable ability with SidM.
In addition to its GEF/GDF activity, SidM also catalyzes the covalent attachment of adenosine monophosphate (AMP) to tyrosine-77 of Rab1, a process known as AMPylation. AMPylated Rab1 is locked in the active conformation because it cannot be inactivated by GTPase–activating proteins (GAPs). AMPylated Rab1 is believed to attract cellular cargo vesicles to the LCV without interference from host proteins that would otherwise deactivate Rab1.
Later during infection, the host factor Rab1 eventually disappears from the LCV. The molecular details underlying such removal have been unclear. The Legionella effector LepB was believed to inactivate Rab1 by triggering GTP hydrolysis, followed by extraction of Rab1 from the LCV membrane by GDI (which binds only to inactive Rab1). However, LepB is unable to inactivate Rab1 in its AMPylated form, leaving open the question as to how L. pneumophila can remove AMPylated Rab1 from its LCV. We discovered that, in addition to encoding the AMPylase SidM, L. pneumophila produces the effector protein SidD, which catalyzes AMP removal from Rab1, a process known as de-AMPylation (or de-adenylylation). Using purified proteins in in vitro de-AMPylation studies, we found that, once AMP is removed from Rab1 by SidD, de-AMPylated Rab1 is accessible for inactivation by GAP proteins such as L. pneumophila LepB. Using indirect immunofluorescence microscopy, we found that L. pneumophila mutants lacking SidD showed a prolonged colocalization with host cell Rab1, demonstrating that SidD is required for the timely removal of Rab1 from LCVs. A similar phenotype was observed in L. pneumophila mutants deficient in the Rab1-GAP LepB, consistent with the notion that both SidD and LepB collaborate to remove Rab1 from LCVs. The identification of L. pneumophila SidD as Rab1 de-AMPylase not only provides the missing link in a complex chain of Rab1 modulation events during L. pneumophila infection but also represents an intriguing example for how prokaryotic and possibly eukaryotic cells may regulate protein function through reversible AMPylation.
Recruitment of host-cell transport vesicles by intracellular L. pneumophila
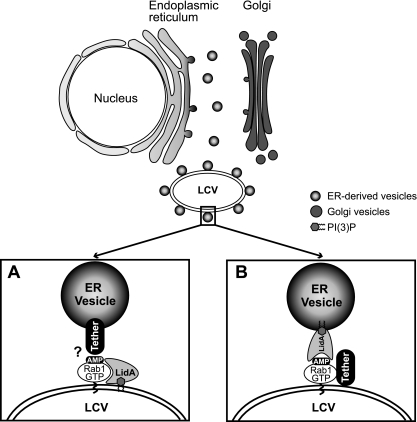
Figure 3. Model(s) of LidA–mediated attachment of ER–derived vesicles to the LCV
(A) LidA translocated to the cytosol of infected host cells localizes to the LCV membrane by binding to AMPylated Rab1 and/or phosphoinosides present on the vacuolar membrane. LidA maintains modified Rab1 in an active conformation and acts as an adaptor protein to mediate the interaction of Rab1 with tethering protein(s) located on the surface of ER–derived vesicles. (B) Upon translocation into the cytosol of infected host cells, LidA localizes to ER vesicles through phosphoinositide binding and mediates docking of these vesicles to the LCV by interacting with AMPylated Rab1 and/or other L. pneumophila effectors present on the LCV that act as tethers.
While the function of SidM, SidD, and LepB has been well characterized, the role of another Rab1-interacting protein from L. pneumophila, the effector protein LidA, is poorly understood. We discovered that LidA binding to Rab1 stabilizes the Rab1-guanosine nucleotide complex, protecting it from inactivation by GTPase-activating proteins (GAPs) and from nucleotide extraction. The protective effect of LidA on the Rab1-guanine nucleotide complex was found to be concentration dependent, consistent with a 1:1 stoichiometry of the LidA-Rab1 complex. The central coiled-coil region of LidA was sufficient for Rab1 binding and to prevent GAP–mediated inactivation or nucleotide extraction from Rab1. In addition, the central region mediated binding to phosphatidylinositol 3-phosphate and other phosphoinositides. When bound to Rab1, LidA interfered with the covalent modification of Rab1 by phosphocholination or AMPylation and also blocked de-AMPylation of Rab1 by SidD and dephosphocholination by Lem3. Based on these findings, we propose a role for LidA in bridging the membrane of the LCV with that of secretory transport vesicles surrounding the LCV (Figure 3).
Interference with the recruitment of Rab1, and thus Rab1–regulated secretory vesicles, to LCVs did not significantly reduce the capability of L. pneumophila to establish replication vacuoles within infected cells. In fact, L. pneumophila mutants lacking either lidA or lidA and sidM were just as proficient as the parental strain in recruiting host-cell vesicles (Figure 4), providing further evidence for the hypothesis that L. pneumophila targets multiple redundant host-cell pathways simultaneously to ensure its intracellular survival.

Click image to enlarge.
Figure 4: Host cell vesicles are recruited to the LCV from multiple trafficking routes.
(Top) Electron micrographs show host vesicles attached to the cytosolic surface of the LCVs at 2 h postinfection of U937 cells with L. pneumophila Lp02 (wild-type), Lp02ΔlidA, or Lp02ΔlidAΔsidM. (Bottom) Magnification of selected regions (boxed) from the upper panel. The arrows indicate vesicles surrounding the LCV.
Additional Funding
- NICHD Director's Challenge Innovation Award
Publications
- Neunuebel MR, Mohammadi S, Jarnik M, Machner MP. Legionella pneumophila LidA affects nucleotide binding and activity of the host GTPase Rab1. J Bacteriol 2012;194:1389-1400.
Collaborators
- Michal Jarnik, PhD, Cell Biology and Metabolism Program, NICHD, Bethesda, MD
Contact
For more information, email machnerm@mail.nih.gov or visit www.nichd.nih.gov/research/atNICHD/Investigators/machner