

You are here: Home > Section on Membrane Biology
Membrane Fusion Mediated by Protein Fusogens

- Leonid V. Chernomordik, PhD, Head, Section on Membrane Biology
- Eugenia Leikina, DVM, Senior Research Assistant
- Kamram Melikov, PhD, Staff Scientist
- Elena Zaitseva, PhD, Research Fellow
- Samristha Sanyal, PhD, Visiting Fellow
- Sung-Tae Yang, PhD, Visiting Fellow
- Santosh K. Verma, PhD, Visiting Fellow
- Ann Hara, BS, Postbaccalaureate Fellow
Disparate membrane remodeling reactions are tightly controlled by protein machinery but also depend on the lipid composition of the membranes. While each kind of protein has its own individual personality, membrane lipid bilayers display rather general properties manifested by their resistance to disruption and bending. Our long-term goal is to understand how, in important cell biology processes, proteins remodel membrane lipid bilayers. We expect that the analysis of the molecular mechanisms of important and diverse membrane rearrangements will shed light on the generality of emerging mechanistic insights. Better understanding of fusion mechanisms will lead to new strategies for quelling diseases involving cell invasion by enveloped viruses, intracellular trafficking, and intercellular fusion. In recent studies, we focused on the membrane-fusion stage of cell entry by dengue virus (DENV), the most prevalent mosquito-borne virus. As do many enveloped viruses such as influenza and hepatitis C viruses, DENV enters host cell via endocytosis. Fusion between the viral envelope and endosomal membrane delivers the viral genome into cytosol. Four distinct serotypes of DENV infect up to 100 million people each year, and an estimated 500,000 people, most of them children, are hospitalized annually with life-threatening dengue haemorrhagic fever/dengue shock syndrome. Currently, there are neither vaccines nor effective therapies for DENV infections.
Recently, we discovered that DENV fusion requires the target membrane to contain anionic lipids such as bis(monoacylglycero)phosphate, a lipid specific to late endosomes, and phosphatidylserine (1). The finding explained why DENV fuses only in late endosomes, while activation of the DENV protein fusogen glycoprotein E is triggered already at a pH characteristic of early endosomes. We proposed that anionic lipid dependence of the DENV fusion machinery protects the virus against premature irreversible restructuring and inactivation and ensures viral fusion in late endosomes, where the virus encounters anionic lipids for the first time during entry. Based on our identification of anionic lipids as a pre-requisite for DENV fusion, we designed several novel assays of DENV entry and used them to explore mechanisms of DENV neutralization by several antibodies.
Structural insights into the fusion-inhibiting activity of a primate antibody against domain I of dengue virus E protein
The envelope glycoprotein E, responsible for target cell recognition and cell entry, consists of three structural domains: DI, DII, and DIII. At one end of the protein E ectodomain is the fusion loop within DII and at the other end is DIII, responsible for host cell binding. An amphipathic stem region connects the ectodomain with transmembrane domains anchored in the viral envelope. Within the acidified endosome, pre-fusion homodimers of protein E dissociate into monomers and insert their fusion loops into the endosomal membrane. E re-assembles into homotrimers with their fusion loops at the tip of a trimeric spike oriented towards the endosomal membrane. Folding back of DIII and stem regions brings membrane-interacting regions of the protein—fusion loops and transmembrane domains—to the same end of the rod-like post-fusion conformation of protein E trimer and, thus, brings the viral and endosomal membranes together. We found that the chimpanzee monoclonal antibody (MAb) 5H2 potently neutralizes DENV-4 by binding to domain I of protein E (2). Crystal structures of Fab 5H2 in complex with DENV-4 sE and isolated recombinant DI provide a snapshot of an intermediate conformation of this domain during protein E refolding between known pre- and post-fusion structures.
The crystal structure of Fab 5H2 bound to DENV-4 protein E shows that antibody binding prevents formation of the fusogenic hairpin conformation of protein E. The finding suggested that 5H2 should block fusion stage of DENV entry. Indeed, we found that the antibody inhibits virus fusion with anionic lipid–containing liposomes. DENV-4 virions whose membranes had been pre-labelled with a self-quenching concentration of the fluorescent lipid DiD were incubated with various concentrations of MAb 5H2 and mixed with liposomes. We then induced fusion of the viral membranes with the liposomes by acidification to pH 5.5, resulting in dilution of DiD in the liposomes and an increase in DiD fluorescence due to de-quenching (Figure 1). MAb 5H2 inhibited lipid mixing in a dose-dependent manner, suggesting that antibody 5H2 neutralizes DENV-4 by blocking membrane fusion within the endosome. Competition ELISA experiments with serum samples from patients convalescing from secondary DENV-4 infections suggest that some antibodies in patient sera bind to epitopes on DI that overlap with that of MAb 5H2. Thus, analysis of 5H2 epitope and neutralization mechanism can help in effective vaccine design to prevent dengue disease.
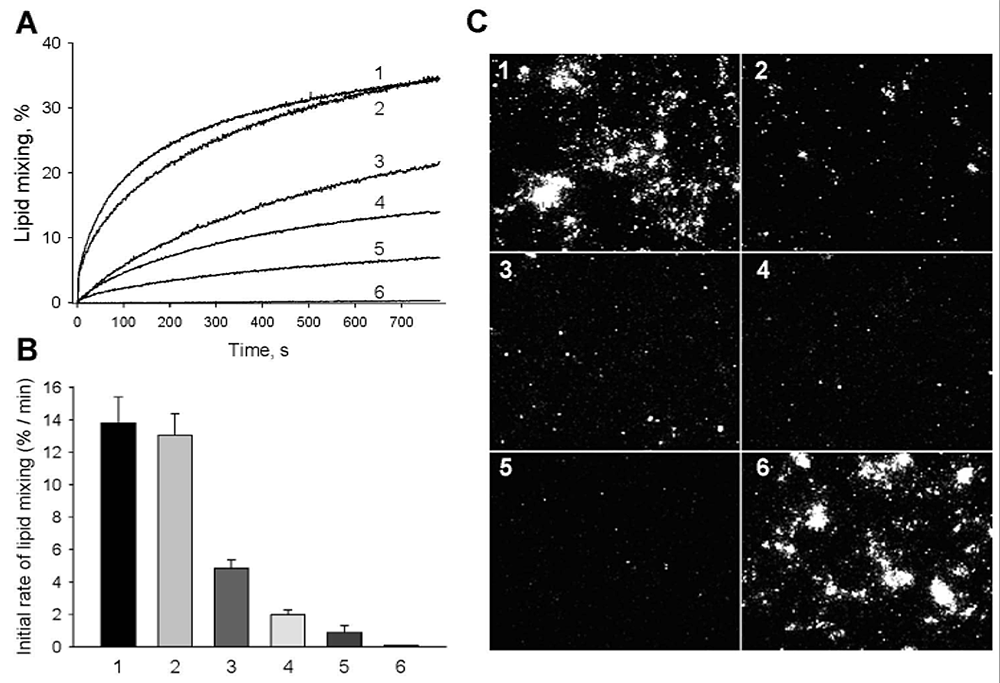
Click image to enlarge.
Figure 1. Neutralizing antibody 5H2 inhibits fusion of DENV-4.
A, B: Fusion between DiD–labeled DENV-4 and liposomes assayed as DiD dequenching. A: Lipid mixing was measured online at pH 5.5, 37°; (1) no antibody; (2) 75 nM nonbinding TBEV Mab 2E6; (3) 30 nM 5H2; (4) 45 nM 5H2; (5) 75 nM 5H2; (6) DEPC–inactivated DENV-4, no antibody. Representative viral fusion curves are from at least three independent experiments. B: MAb 5H2 slows down lipid mixing between DENV-4 and liposomes. Initial rates of lipid mixing observed in the absence of antibody (1), in the presence of 75 nM nonbinding TBEV MAb 2E6 (2), in the presence of 30 (3), 45 (4) and 75 (5) nM 5H2; (6) DEPC–inactivated virus, no antibody. The data are presented as means ± s.d., n≥3. C: 5H2 inhibits fusion of DiD–labeled DENV-4 within the endocytic pathway of cells. Virions preincubated with varying amounts of antibodies were then incubated with BS-C-1 cells grown to near confluency for 30 min at 10°C. Images were taken 40 min after raising the temperature to 37°C to allow internalization of the virus. Lipid mixing was detected as an increase in the cell fluorescence. (1) no antibody; (2) 30 nM 5H2; (3) 45 nM 5H2; and (4) 75 nM 5H2; (5) DEPC–inactivated DENV-4, no antibody; (6) 75 nM nonbinding TBEV MAb 2E6.
Human monoclonal antibodies against dengue virus E protein that target its fusion loop (domain II) and neutralize all four viral serotypes by blocking viral fusion
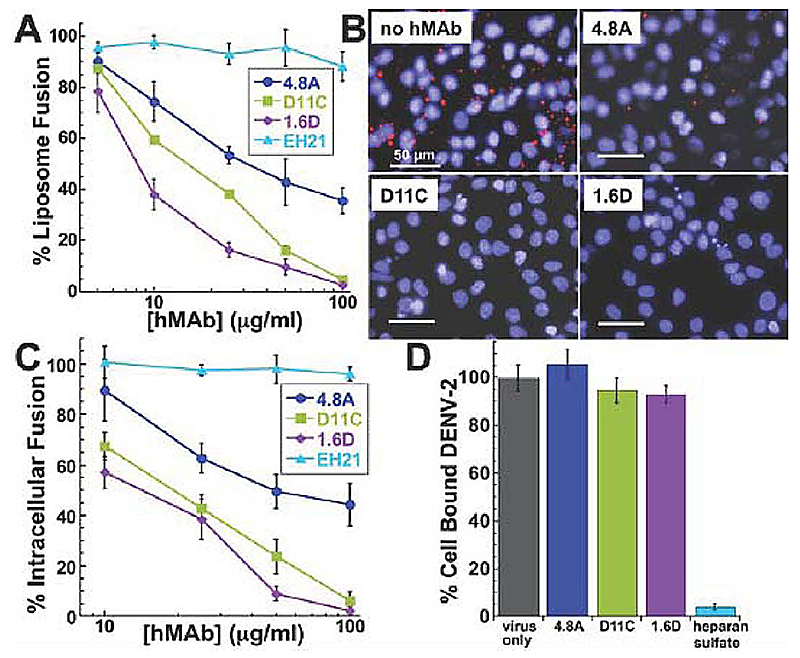
Click image to enlarge.
Figure 2. Mechanism of neutralization by fusion loop antibodies
A: Low pH–activated virus-liposome fusion was measured using fluorescently labeled DENV-2 incubated with hMAbs 4.8A, D11C, and 1.6D (3). The fluorescence signal was normalized to signal generated in the absence of hMAbs to calculate percent liposome fusion.
B: Intracellular fusion of DiD–labeled DENV-2 within endosomes leads to dequenching of DiD. Confluent monolayers of MA104 cells were infected with equivalent amounts of DENV-2 pre-incubated with or without 100 mg/ml hMAbs as indicated. Intracellular structures at the site of fusion events fluoresce red. Cells were counterstained with DAPI to visualize nuclei.
C: Intracellular fusion levels were quantified after incubation of DENV-2 with varying concentrations of hMAbs. EH21 is an irrelevant anti-HIV hMAb. Fluorescence levels were normalized to virus-only controls.
D: Total fluorescence of all bound DENV-2 was quantified by fully dequenching the cells. DENV-2 was incubated with 100 mg/ml of each hMAb. Fluorescence levels were normalized to virus only controls. Heparan sulfate at 10 mg/ml, a known inhibitor of DENV binding, was used as a positive control for binding inhibition.
We screened for broadly cross-reactive and neutralizing human MAbs (hMAbs) from three patients with distinct histories of DENV infection and identified three hMAbs (4.8A, D11C, and 1.6D) that were conformationally sensitive and bound to all four serotypes of DENV equally well, with equilibrium dissociation constants (Kd) in the 10−9 to 10−10 M range (3). Using an ELISA binding-competition assay, we found that hMAbs 4.8A, D11C, and 1.6D bind to overlapping epitopes on E protein. To more precisely define the epitopes for our hMAbs, we identified point mutants that reduce hMAb binding. The residues were located directly within the fusion loop and mapped in close proximity on an E protein structure. All three hMAbs neutralized DENV-1 through 4 to some extent in a dose-dependent manner. Some of the hMAbs were stronger neutralizers than others whereas some neutralized specific serotypes more strongly than others.
Antibodies directed against viral envelope proteins can inhibit viral entry. To determine the details of the mechanism of neutralization, we explored the effects of our antibodies on different stages of viral entry. To investigate whether hMAbs could inhibit DENV-2 fusion, we used an assay that measures fusogenic activity of DENV particles towards liposomes. DENV-2 particles labeled with a self-quenching concentration of DiD were pre-treated with hMAbs prior to co-incubation with liposomes at acidic pH. We monitored virus-liposome fusion (lipid mixing) as an increase in fluorescence reflecting DiD dilution. As expected, no increase in the fluorescence, and thus no lipid mixing, was observed for virions inactivated by the histidine-modifying reagent diethylpyrocarbonate. In contrast to the negative control anti-HIV gp120 hMAb EH21, all three anti-DENV E hMAbs strongly inhibited virus-liposome fusion in a dose-dependent manner (Figure 2A). The relative fusion-inhibiting activity of the hMAbs, with 1.6D being the most potent and 4.8A the least, corresponded to their relative neutralization activity.
Given that virus-liposome fusion relies on random collisions between virions and liposomes rather than on E-mediated virion-liposome binding, the ability of hMAbs 4.8A, D11C, and 1.6D to inhibit fusion between virions and liposomes suggested that viral entry in vivo might also be inhibited at the fusion stage of entry. To test this hypothesis, we directly examined the effects of the antibodies on intracellular fusion of DENV-2 and on the pre-fusion stages of viral entry into rhesus macaque kidney epithelial cells (MA104). For DENV-2 labeled with DiD at a self-quenching concentration, fusion events along the endocytic pathway dilute DiD and thus lead to an increase in fluorescence signal. We quantified the efficiency of intracellular fusion by measuring cell fluorescence with a novel microtiter plate version of the assay described in reference (1). We pre-incubated virions with the antibodies and then applied the virions to the cells at 11°C for 30 minutes to permit binding while holding the virions in a temperature-arrested state. The temperature was then raised to 37°C to allow uptake and fusion of the virions. Fusion of DiD-labeled virus within endosomes leads to dequenching of DiD and the appearance of brightly fluorescent intracellular structures (Figure 2B). Cells were counter-stained with DAPI to visualize the nuclei. All three anti-DENV hMAbs inhibited intracellular fusion in a dose-dependent manner (Figure 2C), corresponding to their relative inhibiting activity in viral neutralization and virus-liposome fusion assays. In contrast, the control anti-HIV gp120 hMAb EH21 did not inhibit intracellular fusion. The results suggest that 4.8A, D11C, and 1.6D directly interfere with the structural transitions required for the virus to fuse to the endosomal membrane.
After measuring the intracellular fusion efficiency, we lysed the cells and fully dequenched the DiD probe in all unfused virions using Triton X-100 to disrupt the viral membranes. The level of unquenched DiD fluorescence was therefore proportional to the total number of cell-associated virions and thus could be used to evaluate the effects of different reagents on virus-cell binding (Figure 2D). As expected, heparan sulfate, known to inhibit DENV binding to cells, dramatically lowered the numbers of cell-associated virions and, consequently, the DiD fluorescence of cell lysates. Pre-incubation of virions with high concentrations of our hMAbs (100 mg/ml, sufficient to profoundly inhibit intracellular fusion) had no effect on DiD fluorescence intensity of cell lysate, indicating that the antibodies do not appreciably affect virus/cell binding. Thus, hMAbs 4.8A, D11C, and 1.6D block viral infection downstream of virus-cell binding at the stage of virus-endosome fusion. Our novel virus-binding and intracellular fusion assay allows parallel quantification of both binding and fusion in the context of viral entry for the same cells and viral particles at physiological temperature and brings to a new level identification of the affected stage of viral entry (binding vs. fusion) in dengue virus neutralization by human antibodies as well as by any other antibodies and antivirals. Characterization of hMAbs targeting the fusion loop region of the E protein may provide new insights into DENV vaccine and therapeutic strategies.
Additional Funding
- NIH's Intramural AIDS Targeted Antiviral Program (IATAP) 2011-2012
Publications
- Zaitseva E, Yang S-T, Melikov K, Pourmal SA, Chernomordik LV. Dengue virus ensures its fusion in late endosomes using compartment-specific lipids. PLoS Pathog 2010;6:e1001131.
- Cockburn JJ, Navarro Sanchez ME, Goncalvez AP, Zaitseva E, Stura EA, Kikuti CM, Duquerroy S, Dussart P, Chernomordik LV, Lai CJ, Rey FA. Structural insights into the neutralization mechanism of a higher primate antibody against dengue virus. EMBO J 2012;31:767-779.
- Costin JM, Zaitseva E, Kahle KM, Nicholson CO, Rowe DK, Graham AS, Bazzone LE, Hogancamp G, Sierra MF, Fong RH, Yang S-T, Lin L, Robinson JE, Doranz BJ, Chernomordik LV, Michael SF, Schieffelin JS, Isern S. Mechanistic study of broadly neutralizing human monoclonal antibodies against dengue virus that target the fusion loop. J Virol 2012;in press.
Collaborators
- Sharon Isern, PhD, Florida Gulf Coast University, Fort Myers, FL
- Michael M. Kozlov, PhD, DHabil, Sackler Faculty of Medicine, Tel Aviv University, Tel Aviv, Israel
- Karl Pfeifer, PhD, Program in Genomics of Differentiation, NICHD, Bethesda, MD
- Félix Rey, PhD, Institut Pasteur, Paris, France
Contact
For more information, email chernoml@mail.nih.gov.